經皮腎交感神經射頻消融術治療中重度心衰的臨床應用
2014-09-12 07:00:06王本文戴啟明馬根山
東南大學學報(醫學版) 2014年5期
王本文,戴啟明,馬根山
(東南大學附屬中大醫院 心內科,江蘇 南京 210009)
心力衰竭是一種復雜的臨床癥狀群,為各種心臟病的嚴重階段。在多種內源性的神經內分泌和細胞因子激活下促進心肌重構,加重心肌損傷和心功能惡化,又進一步激活神經內分泌和細胞因子等,再促使心肌重構,形成惡性循環[1]。早期絕大多數以急性液體潴留為表現的心衰患者對利尿劑治療敏感,但隨著充血性心衰的進展和利尿劑的長期使用交感神經系統會被過度激活,尤其是增加的腎交感神經的活性可使腎血管阻力增加、腎素水平升高和鈉水潴留而致體內液體容量過剩,從而導致利尿劑抵抗的產生,使得心衰控制困難[2- 3]。本研究就是要通過對心衰患者進行雙側腎交感神經消融局部阻滯腎神經的方法,來治療慢性難治性充血性心衰。
1 資料與方法
1.1 一般資料
選擇10例慢性充血性心衰患者,入選標準:紐約心血管協會NYHA心功能分級達Ⅲ~Ⅳ級并伴有難治性容量負荷過重,表現為左室射血分數(LVEF)<40%、全身水腫、腹水、胸腔積液或水腫>2+,并且對常規藥物治療反應差,沒有其他明顯的腎臟病變和腎功能不全的理由(如糖尿病腎病、慢性腎炎和慢性腎臟疾病CKD 4期以上等)。10例患者年齡為47~75歲,平均(62±10)歲,男性8例,女性2例。其中擴張型心肌病5例,缺血性心肌病3例(均經皮冠狀動脈造影證實為三支彌漫性病變),高血壓性心臟病2例。所有患者均經二維超聲心動圖檢查證實為全心擴大,LVEF<40%。所有患者均使用袢利尿劑治療,9例患者使用ACEI類或ARB類藥物治療,7例使用硝酸酯類藥物,3例使用β受體阻滯劑治療。
1.2 治療方法
所有操作均在心內科導管室進行,術前予以鎮靜、鎮痛,消毒后經皮股動脈穿刺建立治療通道后首先進行雙腎動脈造影,排除解剖結構異常后在透視引導下通過引導導管引入射頻消融電極至一側腎動脈遠端,逐漸回撤。在回撤至腎動脈開口的過程中環形旋轉電極頭,沿著動脈壁在不同位置腎動脈內壁上進行4~6點的縱向及環形間斷的局部射頻消融,每個位置的治療時間持續2min,消融的能量為8~12W,每次射頻消融治療點的間隔≥5mm。一側腎動脈完成后再用同樣的方法進行對側腎動脈的治療,消融完畢后再次行雙側腎動脈造影排除腎動脈夾層可能。術后拔除股動脈鞘,壓迫止血,平臥24h。所有患者術后繼續前述抗心衰口服藥物治療。
1.3 觀察指標
觀察患者呼吸困難及水腫癥狀的改善,是否存在假性動脈瘤、心律失常、少尿等手術相關不良并發癥表現。連續檢測患者消融術前和術后的心電、心率、血壓和尿量。在腎神經消融術前24h和術后24h分別抽血檢測血清電解質、血肌酐和血漿神經內分泌激素,包括血清兒茶酚胺、腎素、醛固酮、血管緊張素Ⅱ及腦鈉肽,血漿神經內分泌激素采用放射免疫方法進行檢測。
1.4 統計學處理
本實驗結果使用SPSS 10.0統計軟件處理,實驗數據以均數±標準差表示,組間比較采用單因素方差分析。P<0.05為差異有統計學意義。
1.5 隨訪
通過門診隨訪或電話隨訪的方式了解患者呼吸困難及水腫癥狀的改善,1個月后復查心臟超聲,以了解LVEF變化情況。
2 結 果
接受腎神經消融術后患者的呼吸困難、水腫等心衰癥狀明顯改善,心率、平均動脈壓無明顯變化,患者尿量較術前明顯增加,患者血漿神經內分泌激素包括血清兒茶酚胺、腎素、醛固酮、血管緊張素Ⅱ及腦鈉肽明顯降低,差異均具有統計學意義。術中及術后未發現明顯心律失常、少尿、腎動脈夾層等并發癥。見表1。
表1經皮腎神經消融術術前、術后各項指標的變化
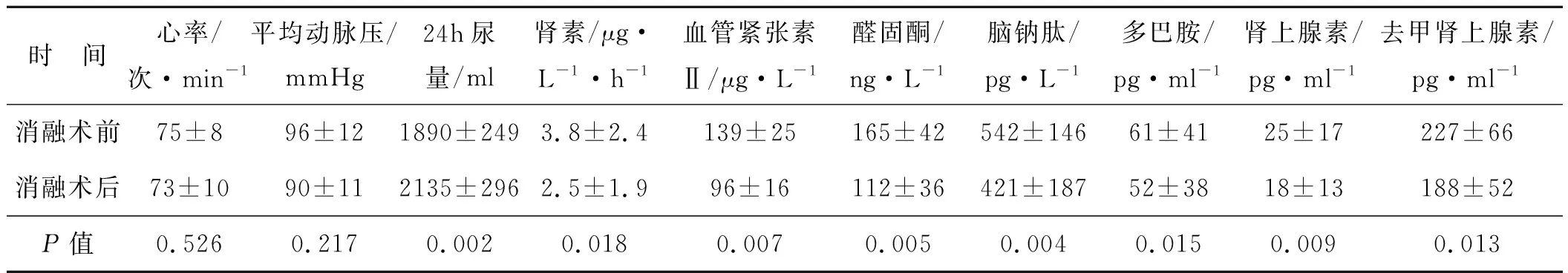
時 間心率/次·min-1平均動脈壓/mmHg24h尿量/ml腎素/μg·L-1·h-1血管緊張素Ⅱ/μg·L-1醛固酮/ng·L-1腦鈉肽/pg·L-1多巴胺/pg·ml-1腎上腺素/pg·ml-1去甲腎上腺素/pg·ml-1消融術前75±896±121890±2493.8±2.4139±25165±42542±14661±4125±17227±66消融術后73±1090±112135±2962.5±1.996±16112±36421±18752±3818±13188±52P值0.5260.2170.0020.0180.0070.0050.0040.015 0.0090.013
術后1個月復查心臟彩超LVEF(43.9%+2.7%)較術前(37.5%±3.6%)相比有明顯提高,P=0.01,差異具有統計學意義,隨訪患者未出現明顯手術相關并發癥,無猝死、惡性心律失常、心衰加重等主要心血管不良事件發生。
3 討 論
導致心衰發生發展的基本機制是心肌重構,心肌重構是由于一系列復雜的分子和細胞機制造成心肌結構、功能和表型的變化。在初始的心肌損傷以后,腎素- 血管緊張素- 醛固酮系統(RAAS)和交感神經系統興奮性增高,多種內源性的神經內分泌和細胞因子激活,其長期、慢性激活促進心肌重構,加重心肌損傷和心功能惡化,又進一步激活神經內分泌和細胞因子等,再促使心肌重構,形成惡性循環。慢性心衰中常有水鈉潴留表現,予以利尿劑治療可緩解心衰患者呼吸困難、水腫等液體量過多表現[4]。隨著心衰的進展和長期利尿劑的使用,將會出現利尿劑不敏感,導致無法清除體內過剩的液體容量,產生利尿劑抵抗,使心衰難以控制[5]。利尿劑抵抗與交感神經過度激活等因素有關,在慢性心衰的晚期常存在交感神經系統的過度激活,尤其是增加的腎交感神經的活性可使腎血管阻力增加、腎素水平升高和鈉水潴留,腎功能下降,利尿劑難以到達正常血藥濃度,從而產生利尿劑抵抗。為治療利尿劑抵抗而住院的患者通常需要靜脈應用影響心肌收縮力的藥物和其他一些有導致心律失常、低血壓和死亡風險的藥物[6- 7]。利尿劑抵抗與增加的死亡率、猝死、泵衰竭死亡獨立相關。
在心衰的治療上,從短期血流動力學、藥理學措施轉為長期的、修復性的策略,目的是改變衰竭心臟的生物學性質。心衰的治療目標不僅僅是改善癥狀、提高生活質量,更重要的是針對心肌重構的機制防止和延緩心肌重構的發展,從而降低心衰的死亡率和住院率。近幾年抑制心衰中過度激活的交感神經活性及RAAS系統活性已經成為心衰治療的重點[8]。歐美和中國心衰指南中均將神經內分泌拮抗藥物,如血管緊張素轉換酶抑制劑和β受體阻滯劑等作為基本治療。同時,在針對心衰液體量過多的治療上也不斷推出了新的藥物及方法。對于難治性心衰國內外目前有部分關于重組人腦利鈉肽及左西孟旦的報道,可見一定成效,但作用時間短、價格昂貴,不能推廣,且對心衰的遠期預后沒有直接影響[9]。 因此,是否能長久有效地降低過度激活的交感神經活性、改善利尿劑抵抗、緩解患者心衰表現、提高生存質量、改善心衰預后成為目前治療慢性心衰的熱點。
近幾年,國外曾進行一項大規模臨床試驗(MOXCON)發現:應用中樞性交感神經抑制劑Moxonidine SR來減低心衰患者的全身交感神經活性,可使全身的去甲腎上腺素水平降低50%,患者心衰癥狀好轉,但是死亡率卻增加,這項試驗因治療組出現較多的死亡病例而提前終止。有報道已證實,局部腎交感神經活性的增加在全身交感神經活性增加中起了非常重要的作用[10]。2006年我院通過對10例慢性心衰伴容量負荷過重的患者采用經皮穿刺注射局麻藥的方法來阻滯一側腎臟交感神經,發現術后患者的尿量、尿鈉排泄率較阻滯前明顯增加,呼吸困難和浮腫明顯減輕,而心率和血壓無明顯變化;患者的血漿神經內分泌激素水平較腎神經阻滯前明顯降低[11]。該方法類似于周圍神經阻滯,其機理主要是通過降低腎臟交感神經的活性減少腎素分泌、擴張腎血管、增加腎血流量,使尿量增加、減少液體潴留、降低心臟的容量負荷、改善患者的心功能和臨床癥狀,以達到治療心衰和利尿劑抵抗目的,但持續時間很短。
因為心衰的發病機制與高血壓的發病機制存在很多相同的機理,所以我們在治療心衰的研究上借鑒高血壓的某些特殊治療方法。近年來,具有突破性的治療頑固性高血壓的方法是使用選擇性的腎交感神經去神經化療法,通過對雙側腎動脈螺旋形多點釋放射頻電磁波能量,透過腎動脈內膜對多數處于外膜的腎交感神經束實施消融,以微創方式選擇性進行交感神經切除,成為了治療頑固性高血壓的很有發展前途的療法,此方法已經大量的臨床試驗證明安全有效。在心衰治療方面,Schlaich等的臨床研究發現患者經腎去交感神經治療后,雙腎局部的去甲腎上腺素釋放水平較術前有明顯下降,術后1年隨訪時其體內的去甲腎上腺素釋放水平較術前下降了42%。同時,隨訪1年后患者心臟核磁共振提示左心室重量較術前下降了15g,心臟體表分布分數較術前亦有所下降。Davies等通過對7例心力衰竭患者術后6個月的隨訪發現,去腎交感神經治療對心力衰竭癥狀有明顯改善,且6min步行距離有顯著增加。以上為本實驗的進行打下了理論及臨床基礎,本實驗的進行已通過了我校醫學院倫理委員會的同意。本實驗通過經皮腎交感神經射頻消融可以長久地減弱交感神經活性,患者的尿量較消融術前明顯增加,呼吸困難和浮腫明顯減輕,而心率和血壓變化不大;患者的血漿神經內分泌激素水平較腎神經消融前有明顯降低,差異具有統計學意義。通過1個月后復查心彩超觀察LVEF變化,期間用藥較術前未有明顯變化,提示左室收縮能力的改善與手術直接相關。這說明應用該療法不但能夠緩解一些伴利尿劑抵抗的難治性心衰的癥狀,而且能夠降低血漿中的神經內分泌激素水平,對患者全身的血流動力學也無不良影響,有望改善患者的預后。但本實驗也存在一定的局限性:首先樣本例數偏少;其次因本實驗為有創性操作,且結合我國實際國情無法做到隨機雙盲,故未設立對照組,僅予以自身前后對照研究;再次,存在其他實驗相關干擾因素,如患者依從性、藥物及其他因素干擾以及患者對手術耐受程度不同導致消融時間長短不一;另外該實驗隨訪時間較短,未能觀察遠期療效及預后。但總而言之,參加實驗的患者術前術后的癥狀及相關檢查數據改善,在一定程度上支持了實驗的可行性及有效性,通過繼續擴大樣本量的實驗可繼續提供相關的有效證據,為廣大中重度心衰患者特別是伴有利尿劑抵抗的人群找到一個合適的治療方法。
[1] PACKER M.Neurohormonal interaction and adaptation in congestive heart failure[J].Circulation,1988,77:721- 730.
[2] ASANO K,DUTEHER D L,PORT S D.Selective downregulation of the angiotensin Ⅱ AT1- receptor subtype in failing human ventrieular myocardium[J].Circulation,1997,95:1193- 1200.
[3] PARATI G,ESLER M.The human sympathetic nervous system:its relevance in hypertension and heart failure[J].Eur Heart J,2012,33:1058- 1066.
[4] BARAJAS L.Innervation of the renal cortex[J].Fed Proc,1978,37:1192- 1201.
[5] BELL- REUSS E,TREVINO D L,GOTTSCHALK C W.Effect of renal sympathetic nerve stimulation on proximal water and sodium reabsorption[J].J Clin Invest,1976,57:1104- 1107.
[6] KIRCHHEIM H,EHMKE H,PERSSON P.Sympathetic modulation of renal hemodynamics,renin release and sodium excretion[J].Klin Wochenschr,1989,67:858- 864.
[7] KON V.Neural control of renal circulation[J].Miner Electrolyte Metab,1989,15:33- 43.
[8] ZANCHETTI A S.Neural regulation of renin release:experimental evidence and clinical implications in arterial hypertension[J].Circulation,1977,56:691- 698.
[9] 趙茂林,羅素紅,衛訓.重組人腦利鈉肽治療難治性心力衰竭療效觀察[J].現代醫學,2012,40(2):218- 220.
[10] YE S,GAMBURD M,MOZAYENI P,et al.A limited renal injury may cause a permanent form of neurogenic hypertension[J].Am J Hypertens,1998,11:723- 728.
[11] 戴啟明,馬根山,馮毅.局部阻滯腎神經治療心力衰竭的臨床應用[J].中國介入心臟病學雜志,2006,14(3):152- 154.
