組蛋白乙酰化與缺血性心肌病
2014-09-12 08:06:12盧圣鋒何蘇云朱冰梅
中國老年學雜志 2014年8期
關鍵詞:研究
黃 艷 盧圣鋒 何蘇云 朱冰梅
(南京中醫藥大學第二臨床醫學院,江蘇 南京 210023)
缺血性心肌病(ICM)是指由于長期心肌缺血導致心肌局限性或彌漫性纖維化,從而產生心臟收縮和(或)舒張功能受損,引起心臟擴大或僵硬、充血性心力衰竭、心律失常等一系列臨床表現的綜合征。隨著全國人口老齡化程度增加, ICM發病率、死亡率急劇上升;在美國約500萬心力衰竭患者中,至少有350萬左心室收縮、舒張功能不全者系ICM所致,已經造成嚴重的社會經濟負擔〔1,2〕。因其機制尚未完全明確,缺乏特異性的治療手段,一直是醫學研究的熱點。表觀遺傳學(Epigenetics)是研究基因核苷酸序列不發生改變的情況下,基因表達了可遺傳的變化的一門遺傳學分支學科;認為遺傳信息主要以DNA甲基化、組蛋白修飾等方式保存,可逆性調節真核基因的表達〔3〕。越來越多的證據表明,ICM是一種與遺傳、環境因素等相關的疾病。組蛋白乙酰化在ICM中的作用越來越受到重視,通過組蛋白乙酰化酶和去乙酰化酶相互作用,在缺血缺氧所致心肌細胞肥大、間質纖維組織增生、廣泛的心肌纖維化中扮演重要角色,這將為ICM的治療提供新的治療靶點,為ICM研究提供新的思路。
1 組蛋白乙酰化修飾調控基因轉錄
1.1組蛋白乙酰化修飾的生物特性 組蛋白乙酰化修飾是基于賴氨酸(Lys)殘基N末端的乙酰化修飾,是目前研究最多最廣泛的組蛋白修飾形式。組蛋白乙酰化和去乙酰化能改變核小體周圍的電荷及染色質構型,當染色質處于相對松弛的狀態,暴露某些轉錄因子的識別位點和結合平臺,利于轉錄因子與DNA結合〔1~4〕。組蛋白末端的共價修飾還構成了獨特的組蛋白密碼,組蛋白的多種修飾方式相互協同作用,構成基因特異性表達的重要基礎。目前研究組蛋白乙酰化修飾的位點較多,主要發生在H3、H4上,不同的乙酰化修飾位點均為激活基因轉錄的作用,組蛋白乙酰化、甲基化修飾共同作用參與轉錄的激活與沉默〔5〕(見表1)。

表1 組蛋白修飾和相關的轉錄調控作用
1.2組蛋白乙酰化修飾相關酶類及相互作用 組蛋白乙酰基轉移酶(HAT)催化組蛋白和非組蛋白乙酰化,是最早被發現的與轉錄有關的組蛋白修飾;而組蛋白去乙酰化酶(HDAC)催化組蛋白去乙酰化。HAT和HDAC共同調節組蛋白乙酰化水平,是組蛋白修飾最重要的方式,也是調控基因表達最主要的驅動力〔6〕,它們各自形成的基因轉錄調控復合物分別稱為輔助激活因子(CoA)和輔助抑制因子(CoR)。前者可使DNA打開,組蛋白乙酰化,激活基因轉錄;后者可使DNA關閉,組蛋白去乙酰化,抑制基因轉錄,兩者共同作用實現基因組整體水平的調控(見圖1〔7〕)。
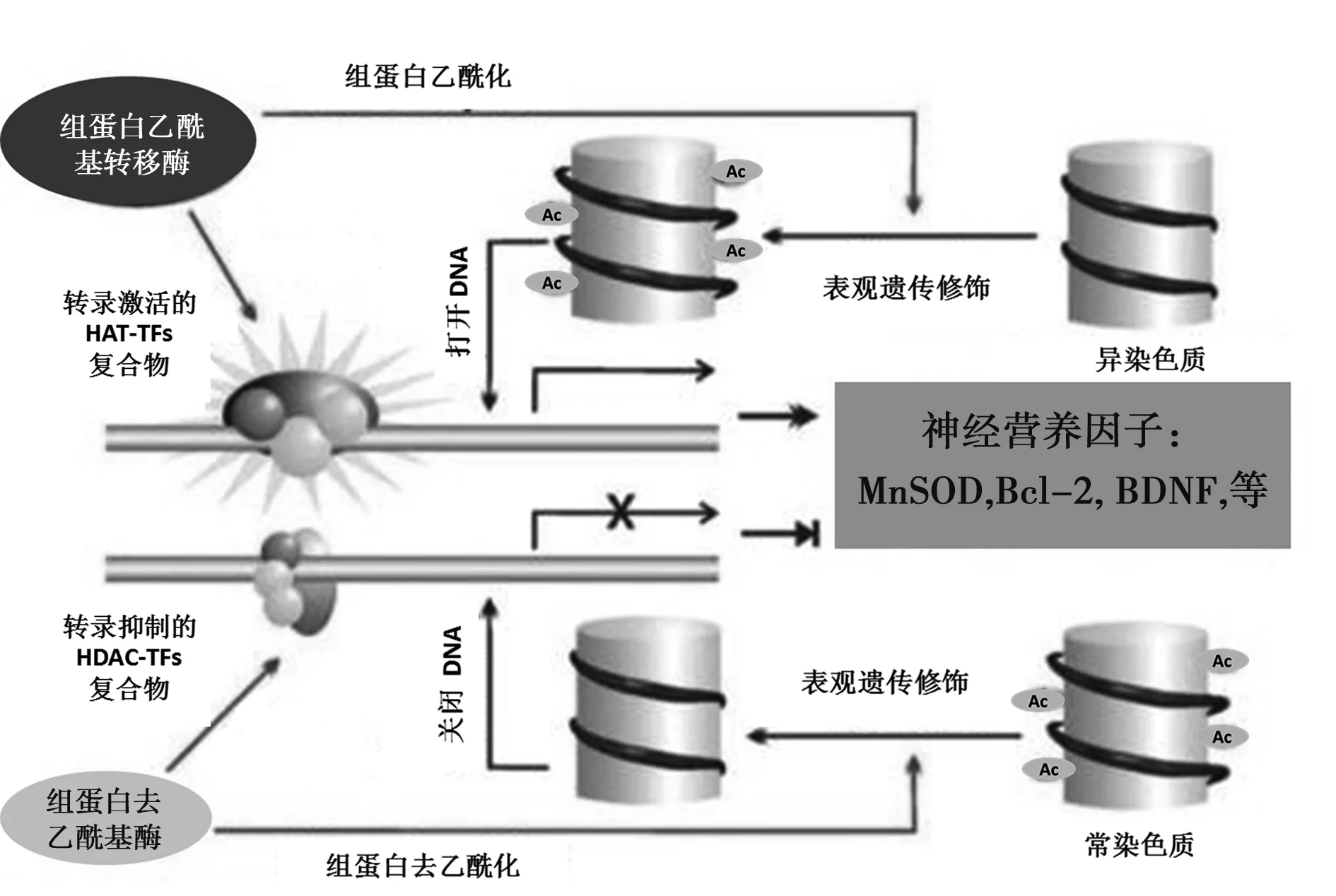
乙酰化轉錄因子活化的結構域招募HATs復合物,打開染色質,催化附近的組蛋白乙酰化,促進基因激活;相反,去乙酰化轉錄因子抑制的結構域招募 HDACs復合物,關閉染色質,催化附近組蛋白去乙酰化,轉錄抑制
HAT的主要功能是將乙酰輔酶A的乙酰基轉移到組蛋白的賴氨酸殘基上,可使核小體組蛋白乙酰化,也可使非組蛋白乙酰化。目前研究發現,20多種HATs已被鑒定具有內源性的A型HATs活性〔8〕(見表2)。
HDAC是一類催化組蛋白的賴氨酸上去乙酰基的酶,在染色質固縮和基因調控上起著關鍵性的作用。目前已知的HDACs按其同源性分為4類(見表3)。
Ⅰ類和Ⅱ類HDACs的活性可以被共同的抑制劑如曲古抑菌素A和n-丁酸等所抑制;Ⅲ類HDACs對這些抑制劑不敏感,但其活性可以被煙酰胺(NAM )抑制〔13,14〕。

表2 HATs的分類和主要作用

表3 HDACs分類及主要作用
2 組蛋白乙酰化修飾與ICM
ICM的病理變化發展過程中,長期慢性缺血缺氧導致心肌細胞能量生成障礙,心肌細胞內Ca2+超載等病理生理改變引發心肌細胞肥大、凋亡、胚胎基因再表達、胞外基質堆積及其組成等變化,導致心肌肥厚、心室重構和心臟的舒縮功能障礙。大量研究表明,組蛋白乙酰化在心肌細胞肥大、心衰等心血管疾病過程中扮演重要角色,直接參與ICM的發生、發展過程〔15,16〕。
2.1組蛋白乙酰化與肌供氧需氧平衡失調 ICM患者,尤其是充血型ICM,往往伴有多支冠狀動脈發生顯著性粥樣硬化性狹窄,導致長期的心肌供氧和需氧之間不平衡,隨著病程進展出現心肌細胞肥大、凋亡。最近的研究表明,HDACs抑制劑能抑制缺氧導致的胚胎基因(GATA)4再表達,減少供需氧失衡導致的血管和心肌損傷,從而保護缺血心肌細胞的功能,提示組蛋白乙酰化在控制心臟的基因表達中具有重要作用〔17〕。此外,HDAC2有可能參與缺氧導致心肌細胞肥大的機制,而HDACs抑制劑可抑制心肌細胞肥大,減少對心臟的缺血性損傷及其梗死面積,減少心功能受損,延遲心衰的發作時間,在心臟重構中發揮重要調節作用〔18,19〕。微小RNA126是HAT/HDAC的缺氧誘導目標,在心肌細胞中的活性可通過激活細胞和促進血管生成途徑對缺血缺氧心肌產生保護作用〔20〕。組蛋白乙酰化參與胚胎基因再表達的調節,抑制心肌細胞肥大,還參與缺血缺氧心肌細胞的激活、心臟血管的生成。
2.2組蛋白乙酰化與心肌細胞能量代謝障礙 ICM病程中伴有心肌細胞能量代謝障礙,心肌缺血可激活交感神經,使心率加快,心肌耗氧量增加,影響肌漿網鈣ATP酶的活性,Ca2+轉入肌漿網內,加速心肌細胞凋亡,進而減弱心肌收縮力。當心肌缺血缺氧時,糖酵解變為能量的主要來源,進而引起心肌能量代謝障礙。在心肌細胞中,叉形頭轉錄因子(FOXO3A)、葡萄糖轉運蛋白(GLUT)4在心肌細胞中參與糖代謝,心肌缺血缺氧引起GLUT4轉位至細胞膜上,引起能量代謝失衡。SIRT1屬于Ⅲ類HDACs,在缺血心肌能量代謝中參與調節內分泌信號,調節FOXO3A、GLUT4,同時調節脂代謝相關的過氧化物酶體增殖物激活受體(PPAR)γ、PPARγ共激活因子PGC-1α,進而調節心肌細胞的能量代謝平衡,在缺血心肌細胞能量代謝中發揮重要作用〔21〕。
2.3組蛋白乙酰化與缺血對心功能和心肌電活動的影響 心肌缺血導致部分心肌細胞壞死,心排血量和每搏量減少,左心室舒張末期容量增加、長期缺血缺氧可致左心室收縮期容量也增加、心臟擴大和心力衰竭。有研究發現,細胞因子的過度表達及連鎖啟動是引發加劇ICM心室舒縮功能障礙的一項重要機制。組蛋白乙酰化修飾中的Ⅰ類HDACs可促進心肌肥厚,如HDAC2可在熱休克蛋白70(HSP70)的誘導下被激活,抑制KLFs樣因子4(KLF4)、多磷酸肌醇-5-磷酸酶F(Inpp5f)等基因促使心肌肥大。相反,Ⅱ類HDACs已被證明是通過抑制心肌特異性轉錄因子如MEF2,GATA4和NFAT發揮抑制心肌肥厚的作用〔22〕。HDAC4、5、7和9 能通過抑制轉錄因子MEF2誘導下游基因的表達,其中HDAC4上結合CaMK的結構域與結合MEF2 的結構域相互交疊, 使得CaMK可以介導MEF2從HDAC上解離, 這表明HDAC-MEF2復合物受Ca2+信號通路中一系列調節因子的控制。但是,現在仍不清楚CaMK/HDAC /MEF2信號通路是否也是心肌細胞分化所必須的〔23〕。美國愛荷華大學研究已證實心肌損傷之后,CaMKⅡ可通過修飾一種特殊的線粒體鈣離子通道來調節鈣離子進入線粒體,其活性增加過大會增加流進線粒體中的鈣離子數量,而這種鈣離子超載觸發細胞死亡〔24〕,心肌功能下降。
ICM病變復雜多樣化,可影響心肌電活動改變心肌細胞膜的通透性,導致胞內鈉水潴留。因心肌細胞內無氧糖酵解的增強,細胞內出現酸中毒、細胞外出現高鉀,影響心室的除極和復極,使心臟沖動的發放和傳導出現異常,引起各種心律失常。HDACs的核易位而發生心肌肥大,導致工作心肌在心肌重構中超極化激活環核苷酸門控4(Hcn4)通道增強子被異位激活,可以使陽離子電流增大,導致致死性心律失常發生,而HDACs抑制劑可激活工作心肌的活動〔25〕。HDAC4從細胞核向細胞質易位可能會引發突觸活動;HDAC3受維甲酸和甲狀腺沉默調解員激素受體(SMRT)和核受體輔阻遏(N-COR)影響,可促使HDACs發生核異位,激活Hcn4通道增強子,引起 K+、Ca2+、Na+等陽離子電流變化〔26,27〕,從而導致各種心律失常。
2.4組蛋白乙酰化與血管內皮功能失調 ICM血管內皮產生和釋放的內源性血管舒張因子一氧化氮(NO)及前列腺素(PGI2)減少,而強有力的縮血管物質內皮素及血管緊張素Ⅱ的分泌增多。內皮祖細胞通過VEGF/PI3K/Akt信號通路分化為成熟的內皮細胞,從而激活HDAC3,HDAC3介導p53蛋白去乙酰化和激活p21。如HDAC3被阻斷,通過PI3K-AKT信號通路下調VCAM-1在EC中的表達,影響血管內皮細胞(EC)的形態與存活〔28〕。Ⅱ類HDACs作為調節心血管疾病的關鍵因素,可抑制MEF2的表達,激活雌激素受體α(ERα)的轉錄,促進病理性心肌重塑。其中HDAC5、9可上調ER靶基因的血管內皮生長因子α(VEGFα)的表達,促進部分梗死區域的血管生成,從而達到保護缺血心肌的作用〔29〕。白藜蘆醇-Sirt1-Foxo1調控通路具有保護缺氧心肌的作用,Sirt1負性轉錄調節因子Foxo1的轉錄活性,其下游基因Bim、p27的表達被調控,抑制血管內皮發展。Sirt1內皮特異過表達可激活一氧化氮合成酶(eNOS)的轉錄,改善血管內皮細胞依賴性主動脈舒張功能,使心肌細胞的細胞周期變長,減少心肌細胞的凋亡〔30〕。
3 小結與展望
ICM是一種常見病、多發病,發病機制涉及心肌供氧需氧平衡失調、心肌細胞能量代謝障礙、缺血對心功能和心肌電活動的影響、血管內皮功能失調等。組蛋白乙酰化在逆轉ICM中扮演重要角色,HDACs廣泛參與疾病的發生發展,調控心肌供需氧平衡、能量代謝、血管內皮功能等多個方面。ICM的基本病因是冠狀動脈動力性和(或)阻力性因素引起的冠狀動脈狹窄或閉塞性病變,是一種受多種因素影響的疾病,如高血壓、高脂血癥、動脈粥樣硬化、糖尿病、肥胖癥等危險因素。Ⅰ類去乙酰化酶HDAC1、2、3均可上調促炎介質的表達,心臟肥大基因和胚胎期基因活化;HDAC8上調可促心肌肥厚標志性基因心房利鈉肽(ANF),參與高血壓發病,促進心肌肥厚發展〔31,32〕。Ⅲ類去乙酰化酶的Sirt1參與高密度脂蛋白(HDL) 的合成,改變膽固醇的運輸,從而降低冠狀動脈粥樣硬化的風險;Sirt1 刺激胰腺分泌胰島素影響血糖代謝,減少了肝臟葡萄糖生成量,有效改善葡萄糖耐量,與胰島素抵抗相關〔33,34〕,可見組蛋白乙酰化修飾與ICM密切相關,值得深入研究。
除此之外,ICM還與環境因素等相關,且同時受飲食、衰老和遺傳等多因素的影響,增加了疾病診療的復雜性。研究表明,組蛋白乙酰化、DNA甲基化、組蛋白甲基化和磷酸化等常常共價修飾,故在研究組蛋白乙酰化修飾與ICM時,也應充分結合其他表觀遺傳修飾調控機制進行研究。通過研究以組蛋白乙酰化為代表的表觀遺傳調控,能很好地闡釋ICM的發病機制,開發更有效的治療用藥。最新研究發現H3K27去甲基化酶UTX在小鼠心臟發育過程中扮演十分重要的角色〔34,35〕,這對表觀遺傳學在心血管疾病研究領域的應用有著積極的推動作用,如HDACs抑制劑將為臨床提供針對性預防和新的治療靶點〔23〕。中醫藥能有效預防和改善ICM,而且以組蛋白乙酰化為代表的表觀遺傳標記物為靶點篩選相應藥物有效成分,也將是一個新的研究方向,值得深入研究探索〔36〕。
4 參考文獻
1王鳴和,王 俊. 缺血性心肌病的研究近況〔J〕. 國際心血管病雜志,2007;34(1):1-5.
2Chaudhry FA, Iskendrin AE. Assessing myocardial viability in ischemic cardiomyopathy 〔J〕. Echocardiography,2005;22 (1):57.
3Schones DE,Zhao KJ.Genome-wide approaches to studying chromatin modifications 〔J〕. Nature Genetics,2008;3(9):179-91.
4Kouzarides T. Chromatin modifications and their function 〔J〕.Cell,2007;128(4):693-705.
5Schübeler D, Mac Alpine DM, Scalzo D,etal. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote 〔J〕. Genes Dev,2004;8(11):1263-71.
6Minucci S, Pelieci PG. Histone deacetylase inhibitors and the promise of epigenetic(and more)treatments for cancer 〔J〕. Nat Rev Cancer,2006;6(1):38-51.
7Saha R,Pahan K.HATs and HDACs in neurodegenaraion:a table of discencerted acetylation homeostasis〔J〕.Cell Death Different,2006;13(4):539-50.
8Eberharter A, Becker PB. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics 〔J〕. EMBO Rep,2002;3(3):224-9.
9Weichert W, RSske A, Niesporek S,etal. Classl histone deacetylase expression has independent prognostic impact inhuman colorectal cancer:specific role of class I histone deacetylasesin vitro and in vivo 〔J〕. Clin Cancer Res,2008;14(6):1669-77.
10Park JH, Kim SH, Choi MC,etal. ClassⅡhistone deacetylases playpivotalrolesin heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors 〔J〕.Biochem Biophys Res Commun,2008;368(2):318-22.
11Michan S, Sinclair D. Siauins in mammals : insights into their biological function 〔J〕. Biochem J,2007;404(1):1-13.
12De Ruijter AJM,Van Gennip AH,Caron HV,etal. Histone deacetylases (HDACs): characterization of the classical HDAC family 〔J〕. Biochem J,2003;370(Pt3):737-49.
13林琴琴,馬 雪,林 蓉.組蛋白去乙酰化酶Sirt1與心血管系統疾病〔J〕.中南醫學科學雜志,2011;39(6):601-6.
14王 穎,王生余,侯春梅,等.組蛋白去乙酰化抑制劑SAHA阻斷MAPK信號通路并誘導HL-60細胞凋亡〔J〕.中國實驗血液學雜志,2007;15(2):267-71.
15Kaneda R, Ueno S, Yamashita Y,etal. Genome-wide screening for target regions of histone deacetylases in cardiomyocytes 〔J〕. Circ Res,2005;97(3):210-8.
16Mano H.Epigenetic abnormalities in cardiac hypertrophy and heart failure 〔J〕. Environ Health Prev Med,2008;13(1):25-9.
17Burch GE, Giles TD, Colcolough HL. Ischemic cardiomyopathy 〔J〕. Am Heart J,1970;79(3):291-2.
18Berry JM, Cao DJ, Rothermel BA,etal. Histone deacetylase inhibition in the treatment of heart disease 〔J〕. Expert Opin Drug Saf,2008;7(1):53-67.
19Shi H, Chen L, Wang H,etal. Synergistic induction of miR-126 by hypoxia and HDAC inhibitors in cardiac myocytes 〔J〕. Biochem Biophys Res Commun,2013;430(2):827-32.
20Pantely GA,Bristow JD.Ischemic cardiomyopathy 〔J〕. Prog Cardiovasc Dis,1984;27(2): 95-114.
21Bush EW, McKinsey TA. Targeting histone deacetylases for heart failure 〔J〕.Expert Opin Ther Targets,2009;13(7):767-84.
22Kemper JK, Choi SE, Kim DH. Sirtuin 1 deacetylase: a key regulator of hepatic lipid metabolism〔J〕. Vitam Horm,2013;91:385-404.
23Olson EN, Backs J, Mc Kinsey TA. Control of cardiac hypertrophy and heart failure by histone acetylation/deacetylation 〔J〕. Novartis Found Symp,2006;274:3-12.
24Joiner ML, Koval OM, Li J,etal. CaMKⅡ determines mitochondrial stress responses in heart 〔J〕. Nature,2012;491(7423):269-73.
25Vedantham V, Evangelista M, Huang Y,etal. Spatiotemporal regulation of an Hcn4 enhancer defines a role for Mef2c and HDACs in cardiac electrical patterning 〔J〕. Dev Biol,2013;373(1):149-62.
26Sando R,Gounko N,Pieraut S. HDAC4 governs a transcriptional program essential for synaptic plasticity and memory 〔J〕. Cell,2012;151(4):821-34.
27Morello F, Perino A, Hirsch E.Phosphoinositide 3-kinase signalling in the vascular system. 〔J〕. Cardiovasc Res,2009;82(2):261-71.
28Rooij E,Fielitz J,Lillian B,etal. Myocyte enhancer factor 2 and Class Ⅱ histone deacetylases control a gender-specific pathway of cardio-protection mediated by the estrogen receptor〔J〕. Circ Res,2010;106(1): 155-65.
29王 偉,陳純娟,傅玉才,等. 白藜蘆醇-Sirt1-Foxo1調控通路對缺氧心肌細胞的保護作用〔J〕. 心臟雜志,2009;21(1):23-8.
30Kee HJ, Kook H. Roles and targets of class Ⅰ and Ⅱa histone deacetylase in cardiac hypertrophy 〔J〕. J Biomed Biotechnol, 2011, 2011:98326.
31曹珊珊,李瑞芳,方偉進,等.組蛋白去乙酰化酶8對腎性高血壓大鼠心肌肥大的影響〔J〕,中國病理生理雜志,2012;28(2):253-7.
32Li X, Zhang S, Blander G,etal. Sirt1 deacetylates and positively regulates the nuclear receptor LXR〔J〕.Mol Cell,2007;28(1):91-106.
33Chen YR, Lai YL, Lin SD,etal. SIRT1 interacts with metabolic transcriptional factors in the pancreas of insulin-resistant and calorie-restricted rats〔J〕. Mol Biol Rep,2013;40(4):3373-80.
34Lee S, Lee JW, Lee SK,UTX.a Histone H3-Lysine 27 demethylase,acts as a critical switch to activate the cardiac developmental program〔J〕.Developmental Cell,2012;22(1): 25-37.
35Wang C, Lee JE, Cho YW,etal. UTX regulates mesoderm differentiation of embryonic stem cells independent of H3K27 demethylase activity〔J〕.Proc Natl Acad Sci USA,2012;109(38):15324-9.
36盧圣鋒,徐 斌,于美玲,等.表觀遺傳學在中醫針灸研究中的應用探討〔J〕.南京中國醫藥大學學報,2013;29(2):105-8.
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
