HPLC測定復方頭孢克洛膠囊含量、含量均勻度及溶出度的研究
2014-06-07 01:59:56張瓊楊利紅蘇蕊
中國醫學創新 2014年17期
張瓊 楊利紅 蘇蕊
復方頭孢克洛膠囊處方源于德國藥物目錄(Pharm Ndex)收載的MUCO Panoral-250,馬丁代爾藥典(Martndale)也收載了頭孢克洛的多組分制劑(Multiingrendient Preparation),Ger( 德 國 ):MUCO Panoral,具有抗感染、止咳祛痰等功效[1-3]。本實驗建立以乙腈-0.025 mol/L磷酸二氫鉀溶液(用磷酸調節pH值至3.0)(22:78)為流動相HPLC方法,同時測定復方制劑中頭孢克洛和溴己新的含量、含量均勻度及溶出度,其專屬性強、靈敏度高、操作簡便,可更有效地控制藥品質量[4-6]。
1 材料
1.1 儀器 島津2010A HCT液相色譜儀(紫外檢測器,工作站),Satorius CP2115天平。
1.2 試藥與試液 頭孢克洛(批號:130481-200904)、鹽酸溴己新(批號:100427-200301),上述對照品均來自中國藥品生物制品檢定所。乙腈:色譜級,其他試劑均為分析純,實驗用水為超純水(Milli-Q Plus超純水系統)制備。
2 方法
2.1 色譜條件 色譜柱:Waters Shield RP C18(4.6 mm×250 mm,5 μm)色譜柱;流動相:乙腈-0.025 mol/L磷酸二氫鉀溶液(用磷酸調節pH值至3.0)(22:78);流速1.0 mL/min,柱溫30 ℃,檢測波長249 nm;進樣量:20 μL。理論板數:以溴己新計為11 215;分離度:頭孢克洛與溴己新的分離度為36.14。該色譜條件下的色譜圖見圖1。

圖1 復方頭孢克洛膠囊HPLC色譜圖
2.2 試驗方法 取本品20粒,精密稱取其內容物(約相當于頭孢克洛50 mg),置100 mL量瓶中,加甲醇30 mL溶解,超聲15 min助溶,冷卻至室溫,加流動相稀釋至刻度,搖勻,濾過,取續濾液作為供試品溶液;取鹽酸溴己新對照品約32 mg,置100 mL量瓶中,加甲醇溶解并稀釋至刻度,搖勻;精密量取上述溶液5 mL,置100 mL量瓶中,加入精密稱取的頭孢克洛對照品約50 mg,加流動相溶解稀釋至刻度,搖勻,作為對照品溶液。精密量取供試品溶液和對照品溶液各20 μL分別注入液相色譜儀,記錄色譜圖,按外標法以峰面積計算,即得。
3 方法學考察
3.1 含量均勻度的測定
3.1.1 含量均勻度線性試驗 精密稱取鹽酸溴己新對照品22.95 mg置10 mL量瓶中,加甲醇溶解并稀釋至刻度,搖勻;精密量取上述溶液2 mL,置50 mL量瓶中,加入精密稱取的頭孢克洛對照品124.41 mg加流動相溶解并稀釋至刻度,搖勻,作為對照品儲備液;精密量取對照品儲備液3、4、5、6、7、9 mL置25 mL量瓶中,加流動相稀釋至刻度,搖勻,制成系列濃度的溶液,按照上述色譜條件依法測定,以濃度與其峰面積進行線性回歸,結果頭孢克洛和鹽酸溴己新回歸方程分別為:Y=19 702X+46 386,r=1.0000;Y=28 912X+1334.2,r=1.0000。線性范圍分別為280.97~842.90 μg/mL和11.00~33.01μg/mL。
3.1.2 精密度試驗 精密量取含量測定項下的對照品溶液20 μL,按照上述色譜條件,連續進樣5次,計算頭孢克洛和鹽酸溴己新峰面積的RSD分別為0.11%和0.24%。試驗結果表明本方法的精密度良好。
3.1.3 重復性試驗 取批號為13020401的復方頭孢克洛膠囊內容物各6份,分別按照“2.2”項下方法進行重復性試驗。結果頭孢克洛和溴己新含量(標示量%,n=6)分別為101.3%和99.6%,RSD分別為0.09%和0.91%。
3.1.4 加樣回收試驗 精密稱取鹽酸溴己新對照品16、20 mg和24 mg分別置25 mL量瓶中,加甲醇溶解并稀釋至刻度,搖勻;精密量取上述溶液2 mL,置10 mL量瓶中,分別加入精密稱取的頭孢克洛40、50 mg和60 mg,加流動相溶解并稀釋至刻度,搖勻,作為儲備液(低、中、高濃度溶液各平行制備3份);另取已測知頭孢克洛和溴己新含量的批號為13020401的復方頭孢克洛膠囊內容物約16 mg(n=9),分別置50 mL量瓶中,精密加入各濃度水平的儲備液2.5 mL,加流動相溶解并稀釋至刻度,搖勻,按上述色譜條件進行加樣回收試驗。結果復方頭孢克洛膠囊中頭孢克洛低、中、高3種濃度的平均回收率(n=3)分別為99.8%(RSD=0.25%)、100.1%(RSD=0.35%)和100.3%(RSD=0.86%),溴己新低、中、高3種濃度的平均回收率(n=3)101.2%(RSD=0.47%)、100.9%(RSD=0.68%)和100.4%(RSD=0.98%)。
3.1.5 溶液的穩定性考察 取含量測定下的供試品溶液,按上述色譜條件,在0、2、4、8、12、25 h依法測定頭孢克洛和溴己新的峰面積,結果RSD(n=6)分別為0.60%和0.53%,表明溶液中頭孢克洛和溴己新在25 h內穩定。
3.1.6 含量均勻度考察 應用本文建立的HPLC含量測定方法,進行兩個批號分別為13020401和131108的復方頭孢克洛膠囊中溴己新含量均勻度檢查。取供試品1粒,置100 mL量瓶中,加甲醇溶解并稀釋至刻度,搖勻,濾過;精密量取續濾液5 mL置25 mL量瓶中,加流動相稀釋至刻度,搖勻,作為供試品溶液,按照含量測定項下的方法測定,同時按復方頭孢克洛膠囊現行標準,以氯仿為溶劑,采用紫外分光光度法在252 nm波長處測定溴己新的含量均勻度[7-8]。兩種方法測定的結果有明顯差異,見表1。

表1 溴己新含量均勻度測定結果
3.2 溶出度測定
3.2.1 溶出實驗 本實驗結合《中國藥典》2010年版二部中頭孢克洛膠囊[9],鹽酸溴己新片與復方頭孢克洛膠囊現行標準YBH04602003中溶出度的方法[8],照《中國藥典》2010年版二部附錄XC第一法,以水900 mL為溶劑,轉速為100 r/min,依法操作,測定了兩批復方頭孢克洛膠囊的溶出曲線,經5、15、30、45、60 min時取溶出液10 mL,用有機膜濾過[9],至少棄去初濾液5 mL,取續濾液作為供試品溶液;另取頭孢克洛與鹽酸溴己新對照品各適量,制成相應濃度對照品溶液,照2.1色譜條件,精密量取供試品溶液和對照品溶液各50 μL分別注入液相色譜儀,記錄色譜圖,按外標法以峰面積計算,計算頭孢克洛和溴己新不同時間的溶出量。溶出實驗結果表明,30 min,6粒膠囊中頭孢克洛平均溶出量達到90%以上,溴己新平均溶出量達到70%以上,6粒膠囊中頭孢克洛與溴己新的溶出曲線見圖2~3。
3.2.2 溶出度線性關系試驗 取鹽酸溴己新對照品20.70 mg置10 mL量瓶中,加乙醇5 mL溶解,加水稀釋至刻度,搖勻;精密量取上述溶液2 mL置25 mL量瓶中,加入精密稱取頭孢克洛對照品125.50 mg,加水溶解并稀釋至刻度,搖勻,作為對照品儲備液。精密量取對照品儲備液3、4、5、8 mL和10 mL置100 mL量瓶中,加溶出介質稀釋至刻度,搖勻,制成系列濃度的溶液,按照2.1色譜條件,精密量取50 μL注入液相色譜儀,記錄色譜圖,以濃度與其峰面積進行線性回歸,結果頭孢克洛和鹽酸溴己新回歸方程分別為:Y=18 166X+3 737 535,r=0.9991;Y=63 769X-30 211,r=0.9991。線性范圍分別為141.71~472.38 μg/mL和4.83~14.53 μg/mL。
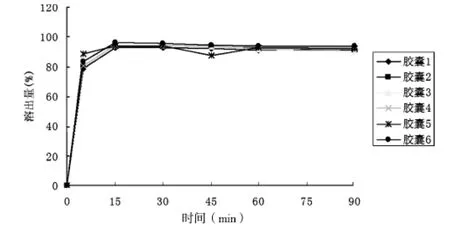
圖2 頭孢克洛溶出曲線圖
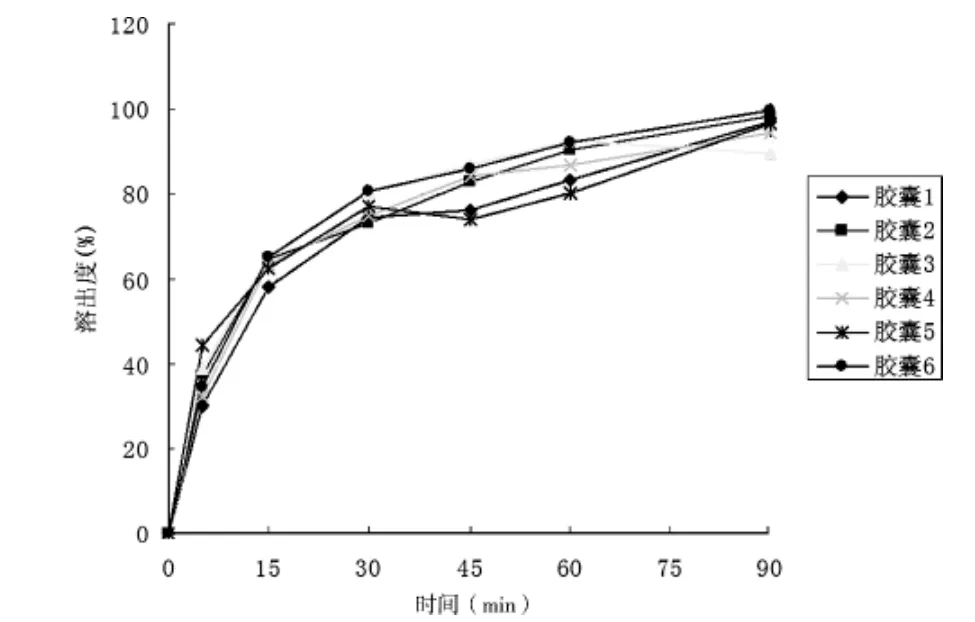
圖3 溴己新溶出曲線圖
3.2.3 加樣回收試驗 精密稱取鹽酸溴己新對照品16 mg、20 mg和24 mg分別置20 mL量瓶中,加適量乙醇(每2 mg加乙醇1 mL)溶解,加水稀釋至刻度,搖勻;精密量取上述溶液2 mL,置25 mL量瓶中,分別加入精密稱取的頭孢克洛50、62和74 mg,加水溶解并稀釋至刻度,搖勻,作為儲備液(低、中、高濃度溶液各平行制備3份);另取已測知頭孢克洛和溴己新含量的批號為13020401的復方頭孢克洛膠囊內容物約16 mg(n=9),分別置100 mL量瓶中,精密加入各濃度水平的儲備液5 mL,加水溶解并稀釋至刻度,搖勻,按2.1色譜條件,精密量取50 μL注入液相色譜儀,進行加樣回收試驗。結果復方頭孢克洛膠囊中頭孢克洛低、中、高3種濃度的平均回收率(n=3)分別為99.6%(RSD=0.22%)、100.0%(RSD=0.50%)和100.2%(RSD=0.64%),溴己新低、中、高3種濃度的平均回收率(n=3)100.8%(RSD=0.72%)、101.3%(RSD=0.88%)和99.9%(RSD=0.47%)。
3.2.4 溶液的穩定性考察 取30 min溶出液作為供試品溶液,按上述色譜條件,在0、2、4、8、12 h依法測定頭孢克洛和溴己新的峰面積,結果RSD(n=5)分別為0.34%和0.55%,表明溶液中頭孢克洛和溴己新在12 h內穩定。
3.2.5 濾膜的吸附作用 取30 min溶出液,分別將原液,無機膜過濾棄去1、2、5 mL初濾液后的溶出液與有機膜過濾棄去1、2、5 mL初濾液后溶出液,精密量取50 μL注入液相色譜儀,記錄色譜圖,結果表明有機膜過濾棄去5 mL初濾液的溶出液中,頭孢克洛與溴己新測定值均與原液測定值的平均偏差小于0.02%,可消除濾膜對頭孢克洛與溴己新的吸附影響。
4 討論
國內現行標準均采用0.005 mol/L四丁基氫氧化銨溶液(用磷酸調節pH值至3.2)-甲醇(62:38)為流動相測定含量,該種試劑對色譜柱損傷大、系統平衡時間長,本文選用磷酸鹽溶液為流動相,大幅減少對色譜柱的損傷,頭孢克洛與溴己新不僅可同時檢出,且分離良好[10-12]。
本文采用HPLC法檢測溴己新的均勻度,成功避免了原標準中有毒揮發性試劑-氯仿的使用,具有專屬性強、靈敏度高、操作簡便、結果準確可靠的特點,同時保護了檢驗人員的健康。
本文方法可同時測定頭孢克洛和溴己新的溶出度,較原標準中僅能檢測頭孢克洛單一組分的溶出量,可進一步提高該產品質量控制。
[1]何其偉,何其莊,鞠萍,等.復方頭孢克洛膠囊處方的優選[J].上海師范大學學報(自然科學版),2002,31(1):74-77.
[2]李新.縣級醫療機構中抗生素合理應用的情況分析[J].中國醫學創新,2011,8(25):132-133.
[3]金曉威,鄭小平,張麗珍,等.靜脈藥物配置中心常見差錯分析及防范[J].中國醫學創新,2011,8(34):111-112.
[4]蔡育紅,鐘慧敏.復方頭孢克洛片分劑量的穩定性試驗[J].廣東藥學院學報,2013,29(3):237-239.
[5]賁春梅.南通市老年康復醫院門診處方抗感染藥物不合理應用420例分析[J].中國醫學創新,2010,7(22):78-79.
[6]吳巧稚,王霆,楊冬玲,等.復方頭孢克洛溴己新及其同類藥研究進展[J].中國藥師,2010,13(5):728-730.
[7]攻麗萍,楊娜,謝元超.HPLC測定復方頭孢克洛顆粒的含量[J].中國藥品標準,2010,11(6):440-442.
[8]國家食品藥品監督管理總局.YBH04602003藥品標準:復方頭孢克洛膠囊[S].2013.
[9]國家藥典委員會.中國藥典[M].第2版.北京:中國醫藥科技出版社,2010:188-190,799-800.
[10]劉衛東,王建成,付慶霞.金剛藤分散片中薯蕷皂苷元HPLC法測定研究[J].中國醫學創新,2012,9(2):3-5.
[11]莫碧芳,黃天國,歐靜.醫院重癥病房抗生素合理應用調查及藥敏分析[J].中國醫學創新,2011,8(8):128-130.
[12]伍良勇,莫蘭芳.HPLC法測定麥角隱亭咖啡因片的溶出度和含量均勻度[J].中國藥品標準,2009,10(1):55-57.
