硼替佐米的合成工藝改進
2014-04-30 07:07:02紀安成韓航王超群張愛華
藥學與臨床研究 2014年2期
紀安成,韓航,王超群,張愛華
1江蘇省藥物研究所,南京 210009;2南京工業大學藥學院,南京 210009
硼替佐米的合成工藝改進
紀安成1,韓航2,王超群2,張愛華1
1江蘇省藥物研究所,南京 210009;2南京工業大學藥學院,南京 210009
對硼替佐米合成工藝進行優化:以異戊醛與R-(+)-苯乙胺為起始原料經縮合、加成、催化氫化和成鹽得到中間體5;另外以L-苯丙氨酸為原料,經酯化、酰化及水解得中間體9;最后中間體5與中間體9經縮合、水解制得目標產物硼替佐米,總收率38.2%(以異戊醛計)。
硼替佐米;多發性骨髓瘤;合成
硼替佐米[1](bortezomib,1,CAS:179324-69-7),商品名為萬珂(Velcade),化學名為[(1R)-3-甲基-1-[[(2S)-1-氧-3-苯基-2-[(吡嗪羧基)氨基]丙基]氨基]丁基]硼酸,是由美國Millennium公司研發的用于治療多發性骨髓瘤(multip lemyeloma,MM)的抗腫瘤藥物,為蛋白酶體抑制劑。該藥于2003年5月在美國上市,并于2005年9月在中國上市。除多發性骨髓瘤外,美國食品藥品管理局(FDA)還于2006年底批準其用于小細胞淋巴瘤;同時在其它類型漿細胞疾病、急性髓系白血病及某些實體瘤的治療上,硼替米佐也被報道有令人矚目的療效。
硼替佐米是有史以來第一個對血液系統及實體惡性腫瘤都具有抗癌效應的蛋白酶體抑制劑,同時可克服化療耐藥性。硼替佐米是一種人工合成的二肽硼酸鹽,曾用代號PS2341,屬于可逆性蛋白酶體抑制劑,可以選擇性地與蛋白酶體活性位點的蘇氨酸結合,抑制蛋白酶體26S亞單位的糜蛋白酶/胰蛋白酶活性,通過抑制特異蛋白質水解,硼替佐米能夠阻斷細胞的多級信號串聯,導致腫瘤細胞死亡。
文獻[2-9]綜述了該藥的合成路線,本文在此基礎上進行整合,改進為一條新的反應路線。首先以異戊醛和R-(+)-苯乙胺為起始原料,在對甲苯磺酸催化下縮合得(R)-N-(3-甲基亞丁基)-1-苯乙胺,其與雙聯頻哪醇硼酸酯在氯化亞銅催化下加成得(R)-3-甲基-N-[(R)-1-苯乙基]-1-胺基-丁基-頻哪醇硼酸酯,接著催化、氫化、成鹽制備得到中間體5,即(R)-3-甲基-1-氨基-丁基-頻哪醇硼酸酯鹽酸鹽;另外再以L-苯丙氨酸為原料,通過酯化,與吡嗪-2-甲酸酰化后水解制備得中間體9,即N-吡嗪羧基-L苯丙氨酸;最后(R)-3-甲基-1-氨基-丁基-頻哪醇硼酸酯鹽酸鹽與N-吡嗪羧基-L苯丙氨酸經縮合、水解反應制得目標產物硼替佐米,總收率38.2%(以異戊醛計)。其合成路線見圖1。
1 實驗部分
1.1 (R)-N-(3-甲基亞丁基)-1-苯乙胺的制備(3)
將異戊醛43 g(0.5 mol,2),R-(+)-苯乙胺66 g(0.55 mol)和對甲苯磺酸一水合物5 g(0.03 mol)投入500 mL甲苯中,加熱回流8 h,冷卻至室溫,依次用飽和碳酸氫鈉水溶液200 mL,水200 mL和飽和氯化鈉溶液200 mL洗滌,有機相直接下一步。
1.2 (R)-3-甲基-N-([R)-1-苯乙基]-1-胺基-丁基-頻哪醇硼酸酯的制備(4)
上步濾液中加入雙聯頻哪醇硼酸酯254 g(1 mol)、氯化亞銅2.5 g(0.025 mol)、補加甲苯300 mL,加熱回流5 h,冷卻至室溫,依次用飽和碳酸氫鈉水溶液200 mL、水200 mL和飽和氯化鈉溶液200 mL洗滌,有機相無水硫酸鈉干燥,過濾,少量甲苯洗滌,濾液濃縮至約500 mL后直接下一步。
1.3 (R)-3-甲基-1-氨基-丁基-頻哪醇硼酸酯鹽酸鹽的制備(5)

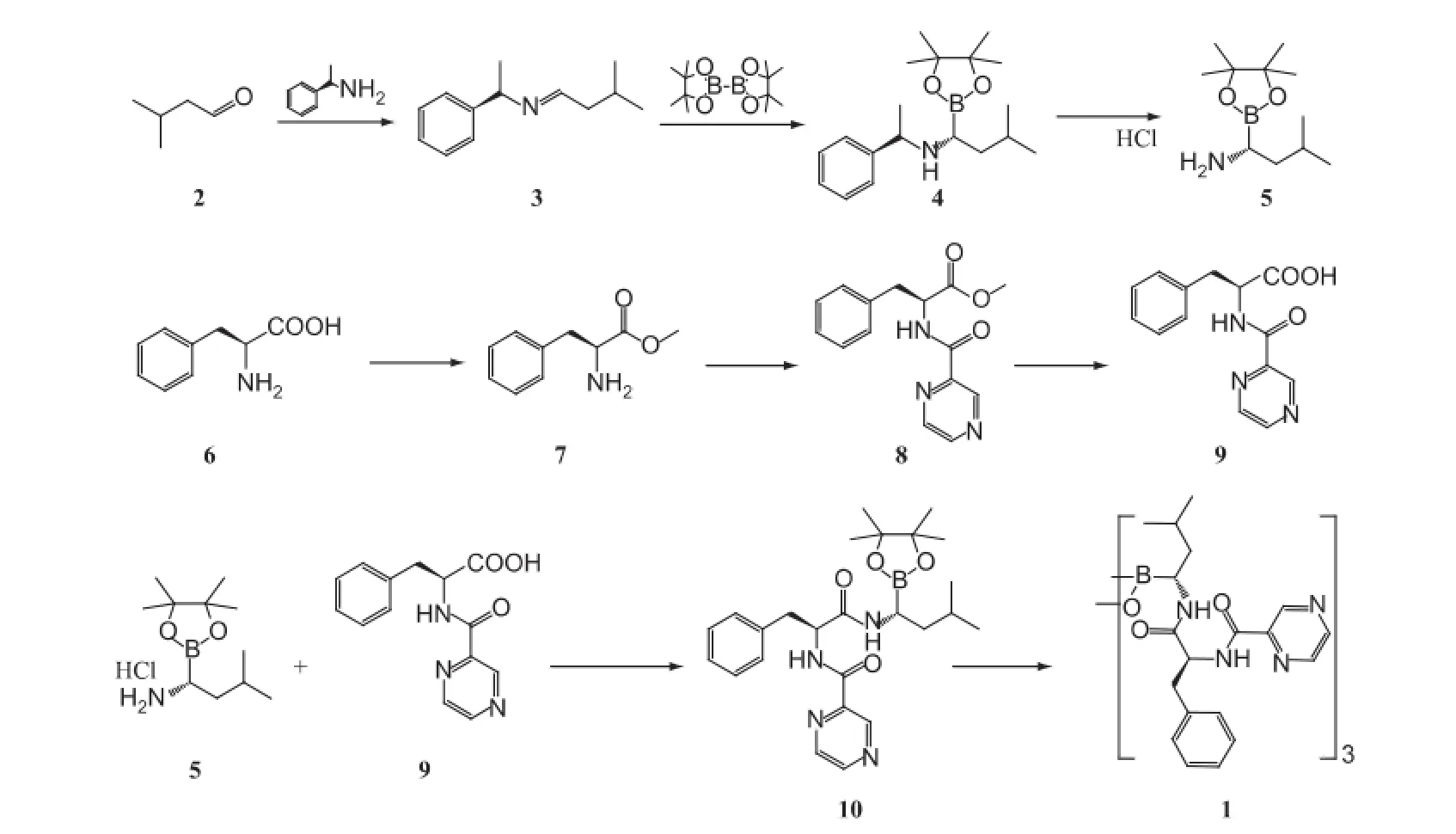
圖1 硼替佐米的合成路線
1.4 L-苯丙氨酸甲酯的制備(7)
反應瓶中加入L-苯丙氨酸165 g(1 mol,6)、二氯甲烷500 mL、98%硫酸10 mL,冰浴冷卻下滴加無水乙醇500 mL(8.6 mol),控制內溫<10℃,滴完后室溫攪拌反應12 h。減壓蒸除溶劑,600 mL乙酸乙酯溶解,分別用2 mol·L-1碳酸氫鉀水液洗滌(200 mL× 3)、水200 mL和飽和氯化鈉溶液200 mL洗滌,有機層無水硫酸鈉干燥。減壓濃縮至干后真空減壓蒸餾,得無色至淡黃綠色液體167.5 g,收率93.6%(文獻[10]收率88.6%)。
1.5 N-吡嗪羧基-L苯丙氨酸甲酯的制備(8)
于反應瓶中加入吡嗪-2-甲酸99.2 g(0.8 mol),N,N-二甲基甲酰胺800 mL,冰浴冷卻至0~5℃,加入N-羥基丁二酰亞胺92 g(0.8 mol),攪拌全溶后分批加入N,N-二環己基碳二亞胺165.4 g(0.8 mol),維持0~5℃攪拌反應20 min后加入L-苯丙氨酸甲酯143.2 g(0.8 mol),于0~5℃攪拌反應10 min,量取110 mL(1 mol)4-甲基嗎啉加入,反應液于0~5℃攪拌反應0.5 h后改室溫反應3 h,薄層示反應完成,過濾,濾液溶于2000 mL乙酸乙酯,分別用水(500 mL×3)、1N鹽酸(500 mL×3)、水500 mL、飽和氯化鈉溶液500mL洗滌,有機相無水硫酸鈉干燥,過濾,濾液濃縮至干,干燥得淡黃色油狀物274.1 g,收率96.2%(文獻[4]收率85.8%)。
1.6 N-吡嗪羧基-L苯丙氨酸的制備(9)
將N-吡嗪羧基-L苯丙氨酸甲酯142.5 g(0.5 mol)加入到600 mL乙醇中,室溫滴加氫氧化鈉22 g(0.55 mol)/水600 mL,保持25~30℃反應1 h,加入5 g活性炭攪拌10 min后降溫至0~5℃,滴加1 mol·L-1的鹽酸至反應液的pH為2~3。控溫0~5℃攪拌反應2 h,抽濾,洗滌,干燥,得白色固體112.1 g,收率82.7%。mp=165~167℃(文獻[4]收率88.7%,mp=166~169℃)。
1.7 [(1R)-3-甲基-1-[[(2S)-1-氧-3-苯基-2-[(吡嗪羧基)氨基]丙基]氨基]丁基]頻哪醇硼酸酯的制備(10)
將(R)-3-甲基-1-氨基-丁基-頻哪醇硼酸酯鹽酸鹽49.9 g(0.2 mol,5)加入反應瓶中,滴加濃氨水調節pH至9~10,二氯甲烷(200 mL×3)提取,提取液分別用水200 mL、飽和氯化鈉溶液200 mL洗滌,有機相無水硫酸鈉干燥,過濾,濾液加入N-吡嗪羧基-L苯丙氨酸81.3 g(0.3 mol,9),2-乙氧基-1-乙氧碳酰基-1,2-二氫喹啉74.1 g(0.2 mol),混合液回流反應20 h,冷卻,濃縮至干得黃色粘稠狀油狀物,直接下一步反應。
1.8 [(1R)-3-甲基-1-[[(2S)-1-氧-3-苯基-2-[(吡嗪羧基)氨基]丙基]氨基]丁基]硼酸(硼替佐米)的合成(1)
上步反應所得油狀物溶于300 mL甲醇中,再加入2-甲基丙烷硼酸40.8 g(0.4 mol)和1N鹽酸300 mL,混合液回流反應8 h,冷卻,加入水300 mL,用二氯甲烷(300 mL×3)提取,提取液2 mol·L-1氫氧化鈉溶液(200 mL×2)洗滌,水層用1 mol·L-1鹽酸調節pH至5~6,后二氯甲烷(200 mL×3)提取,提取液分別用水200 mL與飽和氯化鈉溶液200 mL洗滌,干燥,過濾旋干,干燥,得類白色固體60.3 g。2步總收率78.5%。(文獻[3]總收率70.8%;未見熔點報道;旋光度:=-52°)。所得固體用乙酸乙酯重結晶得白色固體42.6 g,收率70.6%;mp=122~124℃;旋光度:=-52°(C=1,甲醇);含量99.2%(HPLC歸一化法:色譜柱Waters,Symmetry C18柱(250 mm×1.6 mm,5 μm);流動相:乙腈-水-甲醇=30∶70∶0.1;檢測波長:270 nm;流速:25~35 mL·min-1;進樣量:20 μL)。
ESI-MS(m/z):385[M+H]+;1H-NMR(DMSO-d6,500 MHz)數據如下:δH=9.13(s,1H),δH=8.85(dd,3H,J=2 Hz),δH=8.72(s,1H,),δH=7.09(m,5H,J=7 Hz),δH=4.92(dd,1H,J=8.5 Hz),δH=3.08(m,2H,J= 4.5 Hz,9 Hz),δH=2.62(s,1H),δH=1.51(m,1H,J=6.5 Hz),δH=1.26(m,1H,J=6.5 Hz,7 Hz),δH=1.18(m,1H,J=2.5 Hz,7 Hz),δH=0.82(dd,6H,J=2.5 Hz)。
2 討論
(1)在制備化合物5時,以R-(+)-苯乙胺為手性誘導試劑,避免了使用拆分法制備化合物5,從而提高了收率。
(2)在制備化合物10時,以2-乙氧基-1-乙氧碳酰基-1,2-二氫喹啉為催化劑,制備所得化合物10只需簡單蒸出溶劑即可直接用于下一步反應,簡化了操作且不影響產品質量。
本合成工藝避免了高溫、高壓等不利于工業化生產的操作,對各步操作進行了優化,使每步操作更加簡單、安全,同時提高了各步產品質量和收率,避免了頻繁重結晶對收率造成的影響。總收率達到38.2%(以異戊醛計)。
[1] 何東,牛挺.硼替佐米治療多發性骨髓瘤的應用進展[J].華西醫學,2012,27(2):197-202.
[2] 李忠,毛化,鐘靜芬.硼替佐米合成路線圖解[J].中國醫藥工業雜志,2012,43(5):393-5.
[3] 孫江濤,孔立.一種硼替佐米的合成方法:CN101812026[P].2010-08-25.(CA2010,153: 383296).
[4] Li YX,Plesescu M,Sheehan P.Synthesis of four isotopically labeled forms fo a proteasome inhibitor,bortezomib[J].J Labelled Compd Radiopharm,2007,50(5-6):402-6.
[5] Zhu YQ,Zhao X,Zhu XR,et al.Design,synthesis, biological evaluation,and structure-activity relationship (sar)discussion of dipeptidyl boronate proteasome inhibitors,PartⅠ:Comprehensive understanding of the SAR of r-amino acid boronates[J].J Med Chem,2009, 52(14):4192-9.
[6] Pickersgill IF,Bishop J,Koellner C,et al.Synthesis of boronic ester and acid compounds:WO2005097809[P]. 2005-10-20.(CA 2005,143:387386).
[7] Beenen MA,An C,Ellman JA.Asymmetric coppercatalyzed synthesis of α-amino boronate esters from N-tertbutanesulfinyl aldimines[J].J Am Chem Soc,2008, 130(22):6910-1.
[8] Venkata PR,Madinaguda MN,Hyderabad M,et al. Bortezomibandprocessforproducingsame: WO2009036281[P].2009-03-19.(CA 2009,150: 330133).
[9] Ivanov AS,Zhalnina AA,Shishkov SV.A convergent approach to synthesis of vortezomib:the use ofTBTU suppresses racemization in the fragment condensation[J].Tetrahedron,2009,65(34):7105-8.
Synthesis of Bortezomib
JI An-cheng1*,HAN Hang2,WANG Chao-qun2,ZHANG Ai-hua1
1Jiangsu Provincial Institute ofMateria Medica,Nanjing 210009;2School of Pharmaceutical Sciences,Nanjing University of Technology,Nanjing 210009
Intermediate 5 was synthesized from 3-Methyl butanal and(R)-(+)-1-Phenylethylamine by condensation,addition,catalytic hydrogenation and salt-forming.Furthermore,intermediate 9 was synthesized from L-phenylalanine by esterification,acylation and hydrolysis.Finally,the target product,bortezomib was synthesized by condensation of intermediate 5 and intermediate 9 and hydrolysis.The overall yield was 38.2%(based on 3-methyl butanal).
Bortezomib;Multip lemyeloma;Synthesis
R914.5
A
1673-7806(2014)02-121-03
紀安成,男,從事抗腫瘤藥物的設計與合成 E-mail:jac_1982@163.com
2013-12-03
2013-12-24
