UPLC-MS/MS法測定咳速停糖漿中 3 種興奮劑類生物堿
2014-04-11 12:01:17朱平川岑衛健范曉蘇
中成藥 2014年5期
朱平川, 岑衛健, 范曉蘇
(1.廣西大學 亞熱帶農業生物資源保護與利用國家重點實驗室, 廣西 南寧 530004; 2.廣西大學化學化工學院, 廣西 南寧 530004)
UPLC-MS/MS法測定咳速停糖漿中 3 種興奮劑類生物堿
朱平川1, 岑衛健1, 范曉蘇2
(1.廣西大學 亞熱帶農業生物資源保護與利用國家重點實驗室, 廣西 南寧 530004; 2.廣西大學化學化工學院, 廣西 南寧 530004)
目的 建立同時測定咳速停糖漿中鹽酸麻黃堿、鹽酸偽麻黃堿以及磷酸可待因3種有效成分的量的超高效液相色譜-串聯 質 譜 ( UPLC-MS/MS ) 分 析 方 法。 方 法 用 Agilent Zorbax RRHD Eclipse Plus C18( 50 mm ×2.1 mm,1.8 μm) 色譜柱, 以甲醇-0.1%甲酸的水溶液為流動相, 體積流量 0.30 mL/min。 在 電噴霧 電離 ( ESI) 正離子 模式下, 采用的是多重反應監測模式 (MRM) 進行檢測。 結果 鹽酸麻黃堿、 鹽酸偽麻黃堿以及磷酸可待因的線性范圍分別為 0.001 6 ~4.5mg/L、 0.000 8 ~4.8mg/L、 0.004 ~2.0mg/L; 檢出限分別為 0.8 μg/L、 0.4 μg/L、 1.2 μg/L; 3種成分的加樣回收率為 91.1% ~104%; 相對標準偏差均不大于 2.6%。 結論 該方法簡便、 準確、 快速、 靈敏度高,已成功地用于實際的樣品分析。
咳速停糖漿; 生物堿; 超高效液相色譜-串聯質譜 (UPLC-MS/MS)
咳速停糖漿由觀音草、黃精、白尾參、桔梗、 麻黃、罌粟殼、桑白皮等多味苗族藥組成,具有補氣養陰,潤肺止咳,益胃生津的功效。主要用于感冒及慢性支氣管炎引起的咳嗽、咽干、咯痰、氣喘。處方中所含的鹽酸麻黃堿、鹽酸偽麻黃堿、磷酸可待因屬于強成癮性物質,歸屬于興奮劑類物質,但同時也是有效成分。為確保制劑的有效性及安全性,必須控制成分含有量。已有文獻報道對鹽酸麻黃堿、鹽酸偽麻黃堿的測定有高效液相色譜法[1-2]、毛細管電 泳 法[3-4],對 磷 酸 可 待 因 的 檢 測主要是高效液相色譜法[5]、 毛細管電泳法[6]、 液質聯用法[7], 但采用超高效液相色譜串聯質譜法同時測定咳速停糖漿中的鹽酸麻黃堿、鹽酸偽麻黃堿、磷酸可待因這3種物質的分析方法尚未見報道。
色譜-質譜聯用技術具有專屬靈敏、 快速等特點, 已成功用于成分測定[8-10], 更是成分分析 中定性和定量分析的 有 力 工 具[11-15]。 液 相 色 譜-質 譜 聯用技術具有專屬靈敏、快速等特點,是中藥成分分析中定性和定量分析的有力工具[16], 本實驗建立了同時檢測這3種物質含有量的方法,該方法準確、簡便快速,已成功地用于實際樣品的分析。
1 儀器與試藥
1.1 儀 器 1290 Infinity LC System, 6460 Triple Quad LC/MS, 美國 Agilent公 司; Synthesis A10TM超純水系統, 美國 Millipore公司; Eppendorf Centrifuge 5810R臺式高速冷凍離心機,德國 Eppendorf公司; KQ2200DV超聲波清洗器, 昆山市超聲儀器有 限 公 司, ME 215S 電 子 天 平, 德 國 Sartorius公司。
1.2 試劑 甲醇、 乙腈、 甲酸 (色譜純), 美國Fisher公司; 對照品鹽酸麻黃堿、 鹽酸偽麻黃堿、 磷酸可待因,購自中國藥品生物制品檢定所;實驗用水為超純水;樣品咳速停糖漿,貴州白靈制藥有限公司生產, 批號分別為 A20120602、A20120801、A20120607。
2 方法與結果
2.1 色 譜 條 件 采 用 C18色 譜 柱 (2.1 mm × 50 mm, 1.8 μm);以甲醇 (A) -0.1%甲酸水(B) 為流動相, 梯度洗脫 (0 ~2 min, 95%B;2 ~6min, 95%B~93%B; 6 ~8 min, 93%B~ 80%B; 8.0 ~9.0 min, 80%B~10%B; 9.0 ~10 min, 10%B~0%B; 10 ~12min, 0%B~95% B); 體積流量 0.30 mL/min; 進樣體積 1 μL。
2.2 質譜條件 采用電噴霧電離源 (ESI) 正離子模式, 多重反應檢測模式 (MRM模式)。 霧化氣為氮氣, 壓力是 2.8 ×105Pa; 鞘氣為氮氣, 溫度是 360 ℃, 體積流量 12 L/min。干燥氣為氮氣,溫度是 350 ℃, 體積流量 10 L/min。 毛細管電壓4 000 V, 噴嘴電壓 0 V。 3 種目標組分的其他質譜分析參數見表1。

表1 3種成分的部分質譜分析參數Tab.1 MS analysis on parameters of three components
2.3 對照品溶液的配制 準確稱取鹽酸麻黃堿、鹽酸偽麻黃堿、 磷酸可待因對照品各 10.0 mg, 置于10 mL量瓶中, 用甲醇溶解并定容。 對照品貯備液的質量濃度為 1.00 g/L, 于 4 ℃下保存, 使用前用甲醇稀釋到所需的質量濃度。
2.4 樣品溶液的配制 準確量取咳速停糖漿1 m L, 置于 1.5 mL的 EP管中, 12 000 r/min 離心10 min, 將上清液置于另一個 1.5 mL的 EP管中,用甲醇稀釋至 500 mL, 經 0.22 μm的微孔濾膜過濾即可作為樣品溶液。
2.5 線性關系及檢出限 分別吸取各對照品貯備液適量, 以甲醇稀釋配置質量濃度為 0.001 6、0.008、 0.04、 0.2、 1、 2、 4.5 mg/L鹽酸麻黃堿系列溶 液,0.000 8、 0.001 6、 0.008、0.04、0.2、1、 2、 4.8 mg/L鹽酸偽麻黃堿系列溶液, 0.004、0.01、 0.05、 0.1、 0.5、 1、 2 mg/L磷酸可待因系列溶液,在優化的實驗條件下測定,重復3次。以被測組分質量濃度為橫坐標,峰面積的平均值為縱坐標進行線性回歸, 以信噪比 (S/N) 等于 3 為標準, 測得3種待測組分的檢出限, 見表 2, 從表中可看出,各化合物的響應值與質量濃度間均呈現良好的線性關系。

表2 3種組分的線性方程、 相關系數、 線性范圍及檢出限Tab.2 Regression equations, linear ranges, correlation coefficients( r2) and detection lim its of three com ponents
2.6 精密度、 穩定性以及重復性 精密吸取對照品貯備液各 10 μL, 混合稀釋至 10 mL, 配置成含鹽酸麻黃堿、鹽酸偽麻黃堿、磷酸可待因各1 mg/L的對照品混合液, 在優化的實驗條件下連續進 樣 6 次 測 定, 相 對 標 準 偏 差 (RSD) 為0.92% ~1.5%, 結果表明該儀器精密度良好。
取同一批次樣品, 按 “2.4” 項制備樣品溶液, 在優化后的實驗條件下, 分別于 0、 2、4、6、8、 10 h 進樣 測定, 相對標 準偏差 ( RSD) 為1.1% ~1.8%, 結果表明供試品溶液在 10 h 內穩定。
同一批次待測樣品隨機取 6 份,按 “2.4” 項制備樣品溶液,在優化的實驗條件下分析,相對標準偏差 (RSD) 為 1.1% ~2.3%。 結果表明供試樣品重復性良好。
2.7 加樣回收率試驗 分別精密量取已知這 3 種組分含有量的同一批次咳速停糖漿各 1 mL, 共 9份, 每組 3 份, 按 “2.4” 項方法將樣品稀釋 500倍來制備樣品溶液,加入高、中、低3個質量濃度的混合對照品。在最優化實驗條件下進樣測定,3種成分的平均回收率見表3。 在添加3個質量濃度的水平下, 3 種成分的加樣回收率為 91.1% ~104%, RSD均不大于 2.6%, 可以滿足實際樣品的測定要求。

表 3 3 種組分在樣品中的加樣回收率 (n=3)Tab.3 Results of recovery tests of three com ponen ts in sam p les(n=3)
2.8 樣品分析 分別精密稱取不同批次的樣品,按 “2.4” 項制備樣品溶液, 按上述優化好的條件進行樣品測定, 結果見表4。 對照品及樣品的分離情況見圖1, 實驗結果表明, 該方法可用于鹽酸麻黃堿、鹽酸偽麻黃堿、磷酸可待因3種有效成分的同時測定。
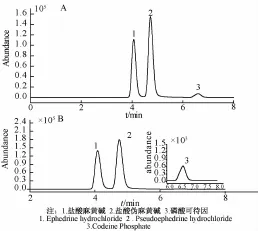
圖1 對照品 (A) 和樣品 (B) 的 MRM色譜圖Fig.1 M ultip le reaction monitoring chromatogram s of three reference substances(A) and sample(B)

表4 樣品測定結果Tab.4 Results of sam p le analysis
3 討論
3.1 色譜條件的優化 本實驗考察了甲醇-水和乙腈-水流動相體系,結果表明使用甲醇-水體系作流動相時靈敏度高于乙腈-水體系,且分離效果更好。在流動相中不添加甲酸時,鹽酸麻黃堿、鹽酸偽麻黃堿兩個物質峰未能達到基線分離,在此基礎上,進一步比較3種物質在不同質量分數甲酸水(0.1%、0.2%、 0.3%、 0.4%) 條 件 下 的 靈 敏度, 結果表明, 0.1%甲酸水最有利于提高靈敏度。3.2 質譜條件的優化 分別對正離子和負離子檢測模式進行了比較,實驗結果表明采用正離子模式檢測這3種物質的檢測靈敏度較高。對選定的各待測物的母離子和定量子離子, 在 MRM模式下進行各種參數的優化, 結果見表1。
4 結論
采用 UPLC-MS/MS 技術建立了同時測定咳速停糖漿中鹽酸麻黃堿、鹽酸偽麻黃堿、磷酸可待因3種成分的分析方法。實驗結果表明,該方法靈敏度高、定量能力強,準確、快速、簡單。該方法的建立有利于進一步完善咳速停糖漿及其類方的質量控制及藥代動力學、藥物療效等相關研究。
參考文獻:
[ 1 ] 韓 杰, 李東輝, 王 鑫.HPLC同時測定感冒軟膠囊中鹽酸麻黃堿、 鹽酸偽麻黃堿及葛根素、 黃芩苷含量[J].中成藥, 2010, 32(6): 963-967.
[2] 張志鵬,朱盛山,李苑新,等.大鼠血漿中麻黃堿偽麻黃堿含量的高效液相色譜法測定[J].時珍國醫國藥, 2012,23(8): 1878-1880.
[3] 曹 娣,馬 婕,宋粉云.毛細管電泳法同時測定小兒清熱止咳口服液中 3 種生物堿類成分的含量[J].中國藥房,2013, 24(12): 1129-1131.
[4] 丁佳佳,李征征,宋粉云.毛細管電泳法測定千柏鼻炎片中鹽酸麻黃堿、鹽酸偽麻黃堿和鹽酸甲基麻黃堿的含量[J].中國實驗方劑學雜志, 2012, 18(10): 120-123.
[5] 王 慧, 毛 睿.復方磷酸可待因糖漿中4種成分的含量測定[ J] .藥物分析雜志, 2009, 29(10) : 1689-1691.
[ 6 ] 李海燕, 木合塔爾, 吐爾洪, 等.CE-ECL法同時測定復方磷酸可待因中三種成分的方法學研究[J].西安交通大學學報: 醫學版, 2009, 30 (6): 769-772.
[ 7 ] 胡紫艷, 陳 云, 孫莉莉, 等.LC-MS/MS 法測定比格犬血漿中的磷酸可待因[J].華西藥學雜志, 2012, 27(4):428-430.
[ 8 ] 楊春林, 李佳峻, 胡 強, 等.超高效液相色譜-串聯質譜法測定發酵調味品中黃曲霉毒素 B1, B2, G1, G2 方法的研究[ J] .中國調味品, 2013, 38(2) : 79-83.
[ 9 ] 陳 勇, 陳澤軍, 周榮清, 等.頂空固相微萃取-氣相色譜-質譜法測定大曲中的揮 發性組分 [ J].中 國調 味品,2013, 38(2): 70-74.
[10] 李 偉.氣相色譜-質譜法測定飲用水中 16 種鄰苯二甲酸酯類化合物[J].食品工業科技, 2011, 32(4): 391-393.
[11] 張亞中.基于超高效液相-四級桿-飛行時間串聯質譜的白花蛇舌草注射液主成分分析[ J].中草藥, 2013, 44(4):829-833.
[12] 趙燕華, 林妮妮, 郭 磊, 等.固相萃取-超高效液相色譜-質譜聯用技術檢測大鼠血漿中 T-2 毒素及其主要代謝產物[ J].分析化學, 2012, 40(12) : 1852-1856.
[13] 侯宏衛, 熊 巍, 郜 娜, 等.固相萃取-高效液相色譜-串聯質譜法同時測定尿液中的 4 種巰基尿酸[J].色譜,2011, 29(1): 31-35.
[14] 張子建, 孫冬曉, 王 婧, 等.LC-MS/MS 法同時測定口炎清 顆 粒 中 8 種 成 分 [ J].中 成 藥, 2013, 35 (5 ):952-956.
[15] 覃 莎, 王 錦, 徐遠金.超高效液相色譜 -串聯質譜同時測定加味左金丸中的 9 種有效成分[J].色譜, 2012, 30(11): 1153-1158.
[16] 金高娃, 章飛芳, 薛興亞, 等.超高效液相色譜在復雜體系中藥分離分析中的應用[J] 世界科學技術: 中醫藥現代化, 2006, 8(3): 106.
Sim ultaneous determ ination of three stim ulant alkaloids in Kesuting Syrupy by UPLC-MS/MS
ZHU Ping-chuan1, CENWei-jian1, FAN Xiao-su2
(1.State Key Laboratory of Conservation and Utilization of Subtropical Agro-bioresources, Guangxi University, Nanning 530004, China; 2.School of Chemistry and Chemical Engineering, GuangxiUniversity, Nanning 530004, China)
AIM A method for simultaneous determination of ephedrine hydrochloride, pseudoephedrine hydrochloride and codeine phosphate in Kesuting Syrupy was established by ultra performance liquid chromatography with tandem mass spectrometry( UPLC-MS/MS) .M ETHODS The UPLC separation was performed on a Zorbax RRHD Eclipse Plus C18column ( 2.1 mm× 50 mm, 1.8 μm) by using methanol and water which contained 0.1%formic acid asmobile phase with the gradient elution at a flow rate of 0.30 m L/min.The analytes were detected by tandem mass spectrometry under the positive ionmode with the ESIsource and carried out in the multiple reaction monitoring( MRM) mode.RESULTS Under the optimum conditions, the calibration curves were linear in the range of 0.001 6-4.5 mg/L for ephedrine hydrochloride, 0.000 8-4.8 mg/L for pseudoephedrine hydrochloride, 0.004-2.0 mg/L for codeine phosphate.The detection limitswere 0.8, 0.4, 1.2 μg/L, respectively.The average recoveries of the three effective componentswere between 91.1%and 104%with all relative standard deviations notmore than 2.6%.CONCLUSION The developedmethod is simple, rapid and accurate, and suitable for the quality control of the three components in Kesuting Syrupy.
Kesuting Syrupy; alkaloid; ultra performance liquid chromatography-tandem mass spectrometry(UPLC-MS/MS)
R927.2
: A
: 1001-1528(2014)05-0970-04
10.3969/j.issn.1001-1528.2014.05.019
2013-08-19
廣西自然科學基金項目 (桂科自0832034); 廣西大學科研基金項目 (DD703003)
朱平川 (1982—) , 女, 實驗 師, 從 事 色 譜、 質 譜 及 其 聯 用 方 法 的 研 究。 Tel: ( 0771 ) 3237743, E-mail: zhupingchuan12@ 163.com
