基質固相分散萃取-氣相色譜-串聯質譜法同時測定蔬菜中195種農藥殘留
2014-03-08 09:18:08盧俊文張朋杰李擁軍謝建軍儲大可
食品科學 2014年24期
關鍵詞:檢測
李 蓉,盧俊文,楊 芳,張朋杰,李擁軍,謝建軍,儲大可
(1.中山出入境檢驗檢疫局,廣東 中山 528403;2.中山市農產品質量監督檢驗所,廣東 中山 528400;3.廣東出入境檢驗檢疫局,廣東 廣州 510623;4.廣東藥學院,廣東 中山 528400)
基質固相分散萃取-氣相色譜-串聯質譜法同時測定蔬菜中195種農藥殘留
李 蓉1,盧俊文1,楊 芳1,張朋杰1,李擁軍2,謝建軍3,儲大可4
(1.中山出入境檢驗檢疫局,廣東 中山 528403;2.中山市農產品質量監督檢驗所,廣東 中山 528400;3.廣東出入境檢驗檢疫局,廣東 廣州 510623;4.廣東藥學院,廣東 中山 528400)
目的:建立一種蔬菜中195 種農藥的氣相色譜-質譜檢測方法。方法:樣品用乙酸乙酯勻漿提取后,經基質固相分散萃取法凈化,氣相色譜-質譜儀的選擇反應監測模式-進行測定,內標法定量。結果:在0.01~1.00 mg/L質量濃度范圍內,所有農藥的線性相關系數均大于0.99,方法的定量限范圍為1.9~25.5 μg/kg。分別在菜心、生菜、黃瓜、芹菜、土豆等蔬菜中進行3 個添加水平(10、20、100 μg/kg)實驗,平均回收率為37.6%~136.7%,相對標準偏差為0.2%~15.3%。結論:該方法快速、靈敏、準確、高通量,具有良好的回收率和穩定性,較好地解決了樣品本底成分復雜帶來的基質干擾,適用于蔬菜中195種農藥殘留量的快速測定。
基質固相分散萃取;氣相色譜-質譜;農藥殘留;蔬菜
在《中國居民膳食指南》中,蔬菜的建議攝入量占據了相當高的比例。農藥在蔬菜的生長過程中發揮了重要作用,但因施用人員不注意使用的間隔時間,會造成上市蔬菜農藥殘留超標。近年來,世界各國對農產品中限制檢出農藥殘留的種類迅速擴大,限量標準的要求更加嚴格。2013年3月1日,我國開始實施GB 2763—2012《食品中農藥最大殘留限量》,規定了食品中322種農藥2293項最大殘留限量,由此對檢測技術的要求進一步提高。
目前農藥殘留檢測手段有氣相色譜技術[1-3]、液相色譜技術[4]、氣相色譜-質譜聯用技術[5-7]、液相色譜-串聯質譜(liquid chromatography-tandem mass spectrometry,LC-MS-MS)技術[8-10]、氣相色譜-串聯質譜(gas chromatography-tandem mass spectrometry,GC-MS-MS)技術[11-13]和酶聯免疫吸附法[14]。GC和LC由于受檢測器限制,一般只能對一類農藥進行檢測,依靠保留時間定性,易出現干擾現象,準確性相對較差;GC-MS技術在分析較高沸點或低相對分子質量的農藥時,易受基質成分和柱流失物等因素影響而導致靈敏度較低或發生干擾現象;LC-MS-MS技術靈敏度高,抗干擾能力強,但儀器昂貴維護成本高,目前難以推廣普及;酶聯免疫吸附法法對于多種成分的分析存在局限性。
本實驗利用分散固相萃取(dispersive solid-phase extraction,DSPE)[15-18]-GC-MS-MS技術,建立了同時測定蔬菜中195 種農藥殘留的方法,前處理簡單,試劑毒性小,消除了GC-MS技術在分析蔬菜尤其是復雜基質樣品中農藥殘留時極易出現的干擾現象,大幅降低了檢測成本,利于推廣。本法測定的農藥涵蓋了有機磷類、有機硫代磷酸酯類和有機氯類殺蟲劑,酰胺類殺螨劑,三唑類和有機氮保護性殺菌劑,苯胺基嘧啶類、酰胺類除草劑及殺蟲劑前體等多種農藥,使用選擇反應監測模式和內標法定量,提高了分析的靈敏度和準確性,適用于蔬菜中農藥多殘留的快速篩查和確證檢測。
1 材料與方法
1.1 材料與試劑
乙酸乙酯(色譜純) 美國Merck公司;Cleanert N-丙基乙二胺(primary secondary amine,PSA)天津博納艾杰爾科技有限公司;C18(分析純) 德國CNW公司;無水硫酸鎂、醋酸鈉、無水硫酸鈉(均為分析純) 廣州化學試劑廠;195種農藥標準品(其中三唑磷的純度為81.0%,其余標準品的純度≥95.0%)、磷酸三苯酯(triphenyl phosphate,TPP) 德國Dr.Ehrenstorfer公司。
1.2 儀器與設備
TSQ Quantum XLS系列三重四極桿質譜儀及其工作站 美國Thermo Fisher Scientific公司;2-16K臺式離心機 德國Sartorius-Sigma公司;EYELA MMV-1000W振蕩器 日本東京理化;渦旋混合器 德國IKA公司;TruboVapII全自動濃縮儀 美國Caliper公司;超純水系統 美國Millipore公司。
1.3 方法
1.3.1 標準溶液的配制
各種農藥單標儲備液的配制:分別準確稱取各農藥標準品10 mg(精確至0.000 1 g),用乙酸乙酯溶解定容至10 mL,配制成1 g/L的標準儲備液,儲存于-18℃冰箱中備用。
混合標準儲備液的配制:各取0.5 mL上述標準儲備液,用乙酸乙酯定容至100 mL,即配制成各農藥標準質量濃度為5 μg/mL的標準混合儲備液。
混合標準工作液的配制:精密量取5.0 mL混合標準儲備液,用乙酸乙酯定容至25 mL,配制成各農藥標準質量濃度為1 μg/mL的標準混合工作液(為了避免多種農藥共存對各自的穩定性產生影響,混合標液均臨用現配)。
TPP內標溶液的配制:準確稱取12.5 mg,用乙酸乙酯溶解并定容至25 mL;吸取上述溶液1.0 mL,用乙酸乙酯定容至25 mL,配制成20 μg/mL的內標溶液。
1.3.2 GC條件
色譜柱:TR-PESTICIDE(30 m×0.25 mm,0.25 μm);進樣方式:程序升溫不分流進樣,程序升溫汽化進樣口:起始溫度50℃,以8℃/min升至280 ℃;載氣:He,99.999%;柱溫采用程序升溫,初始溫度50℃,保持1 min,以30℃/min升至160℃,以5℃/min升至300℃,再以5℃/min升至310℃,保持3 min;柱流量1.3 mL/min;進樣量:1 μL。
1.3.3 MS-MS條件
離子源溫度:240℃;電離模式:電子電離源;質譜傳輸線溫度280 ℃;溶劑延遲時間4.5 min;碰撞氣(Ar)壓力:1.5 mTorr;分析模式:定時選擇性反應監測模式;定量方法:峰面積內標法定量。
1.3.4 樣品提取與凈化
稱取樣品10 g(精確到0.01 g)于50 mL離心管中,加入6 g MgSO4和2 g醋酸鈉,準確加入50.0 μL TPP內標溶液,渦旋1 min;加入約5g(視樣品含水率增減)經450℃灼燒過的無水硫酸鈉,再準確加入20.0 mL乙酸乙酯,勻漿提取1 min,再置入振蕩器振搖30 min,以4500 r/min離心5 min;準確吸取10 mL上清液上全自動濃縮儀(水浴溫度調節到40℃),氮吹濃縮至約1 mL,定容至1.0 mL;將定容后的溶液加入150 mg MgSO4、50 mg PSA(根據需要再加50 mg C18),渦旋3 min,以轉速為10000 r/min離心10 min,取上清液待GC-MS-MS分析。
2 結果與分析
2.1 前處理方法優化
2.1.1 提取溶劑的選擇
常用的萃取溶劑有乙腈、乙酸乙酯、丙酮、甲苯、二氯甲烷等。因甲苯和二氯甲烷毒性強,對實驗者傷害大,本實驗中盡量避免將其作為提取溶劑。分別采用乙腈、乙酸乙酯、丙酮作為提取溶劑,乙腈的回收率為60.7%~125.1%,乙酸乙酯的回收率為53.2%~136.6%,丙酮的回收率為50.6%~150.9%。實驗表明,乙腈的提取效果最好,乙酸乙酯次之,丙酮最差,但考慮到乙腈的毒性比乙酸乙酯大,所以選擇低毒性的乙酸乙酯作為提取溶劑。
2.1.2 提取液的pH值對測定結果的影響
在提取溶劑中加入少量弱酸及弱酸鹽可以形成緩沖體系,降低基質溶液酸堿性的影響,從而有效提高農藥的提取率。對比加入不同量的醋酸鈉(0、0.5、1、2、3 g)對提取效率的影響,發現加入2 g時絕大部分農藥的回收率相對最高。 因此最后確定在提取溶劑中加入2 g醋酸鈉。
2.1.3 前處理方法的選擇
蔬菜是一類基質復雜的樣品,本實驗嘗試采用如液-液萃取法、勻漿法、加速溶劑提取法、凝膠滲透萃取法、固相萃取法等對樣品進行前處理操作,部分復雜基質難以消除,且不同前處理方法會造成不同農藥成分的回收率偏低,影響檢測結果。采用DSPE法對蔬菜樣品進行前處理,方法快速簡便、試劑消耗量小,且能有效去除復雜基質,大大減少了測定干擾。以菜心為例,幾種前處理方法的比較見圖1。

圖1 加速溶劑(A)、氨基固相萃取(B)和DSPE(C)法空白基質圖Fig.1 Total ion chromatogram of blank matrix by different extraction methods
由圖1可見,采用DSPE法凈化后的空白基質(圖1C)雜峰的干擾大大減少,能除去蔬菜中絕大部分的復雜基質,達到理想的凈化效果。
2.2 色譜條件優化
通過對進樣口、3種相同規格不同極性色譜柱(DB-1701、DB-5和TR-PESTICIDE)的對比使用,以及對柱流量和升溫程序的調整,確定了1.3.2節的GC條件。
2.3 質譜條件優化
本方法選擇SRM模式下的2對離子對進行各農藥的分析,其中1對離子對用于定量分析。為了獲得最佳的質譜條件以保證定性定量的準確性,對各項質譜參數進行了優化。首先采用單一標準品進樣,全掃描方式測定其保留時間和選擇碎片離子,確定母離子,再采用產物離子掃描方式通過優化碰撞能量獲得豐度比較高產物離子,并確定被測物SRM離子對,最后采用SRM模式對待測物進行定性定量分析。經過條件優化得到了理想的質譜條件和分離效果,195 種農殘組分優化后SRM模式下的GCMS-MS條件見表1。圖2為優化條件下0.05 mg/L基質加標工作液的總離子流色譜圖。
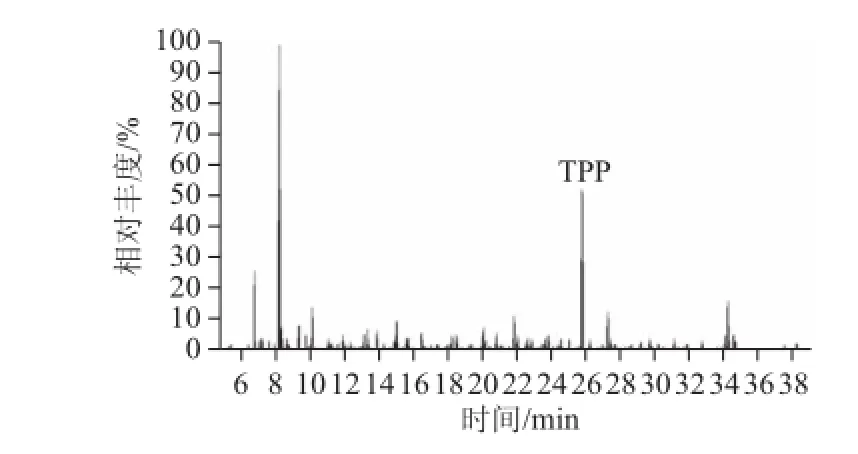
圖2 基質加標工作液(質量濃度0.05 mg/L)的總離子流圖Fig.2 Total ion chromatogram of blank matrix spiked at 0.05 mg/L
2.4 線性范圍與定量限
使用空白的蔬菜樣品配制系列質量濃度的基質加標工作曲線(0.01~1.00 mg/L),在1.3.2、1.3.3節選定的GC、MS-MS條件下進樣測定,以標準工作溶液中各農藥的質量濃度(x,mg/L)為橫坐標,定量離子提取色譜峰面積(y)為縱坐標繪制標準曲線,獲得各農藥的線性方程,相關系數均大于0.99。在空白樣品中添加低質量濃度的混合標準溶液,按1.3.4節方法進行前處理后上機檢測,按10倍信噪比得到各農藥的定量限為1.9~25.5 μg/kg(表1)。
2.5 回收率與精密度
本實驗對菜心、生菜、黃瓜、芹菜、土豆等幾種蔬菜加標進行回收率和精密度測定。稱取各空白樣品10 g各18 份,在其中分別添加10、20 μg/kg和100 μg/kg添加水平,按1.3.4節進行前處理,在1.3.2、1.3.3節條件對樣品進行分析,測定后計算回收率和相對標準偏差。結果顯示,方法的加標平均回收率為37.6%~136.7%,相對標準偏差為0.2%~15.3%,菜心的加標回收實驗結果見表1。

表1 優化后的多反應監測模式下的氣相色譜-串聯質譜儀的條件、加標回收率、精密度和定量限TTaabblle 1 Optimized conditions in MRM mode for GC-MS-MS, recoveries, precisions (RSDs) and limits of quantification (LOQs)
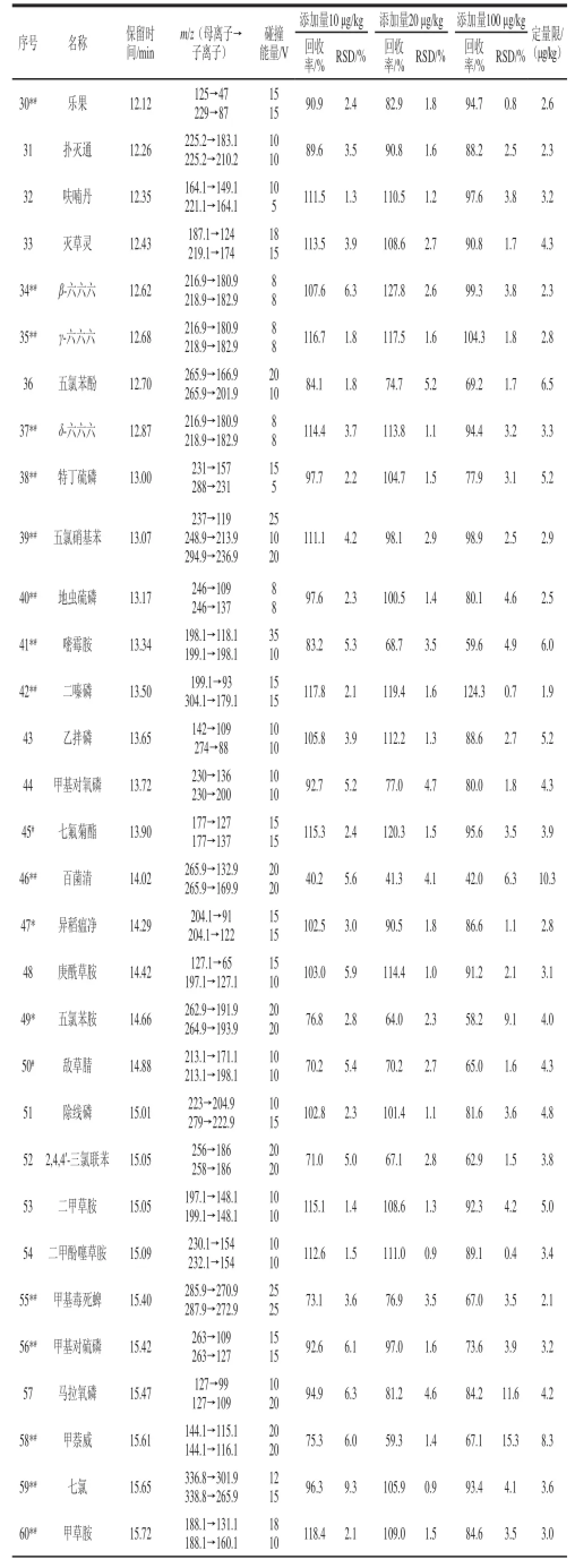
續表1

續表1

續表1
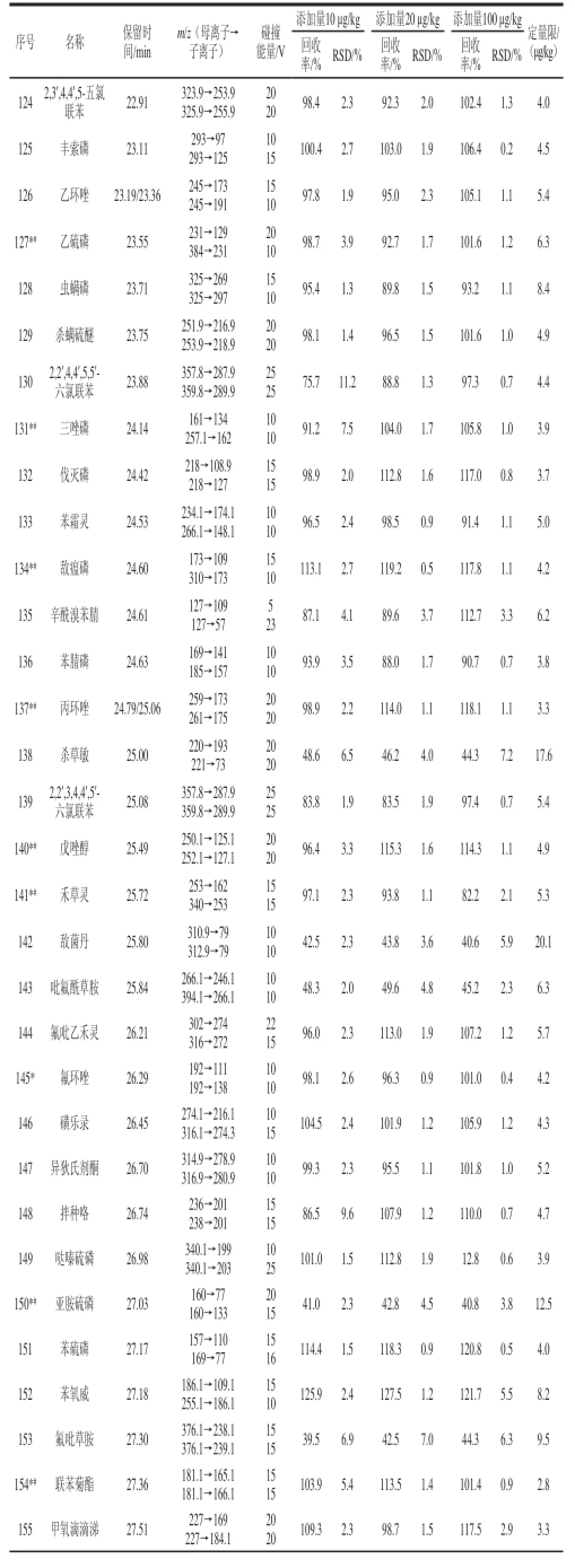
續表1
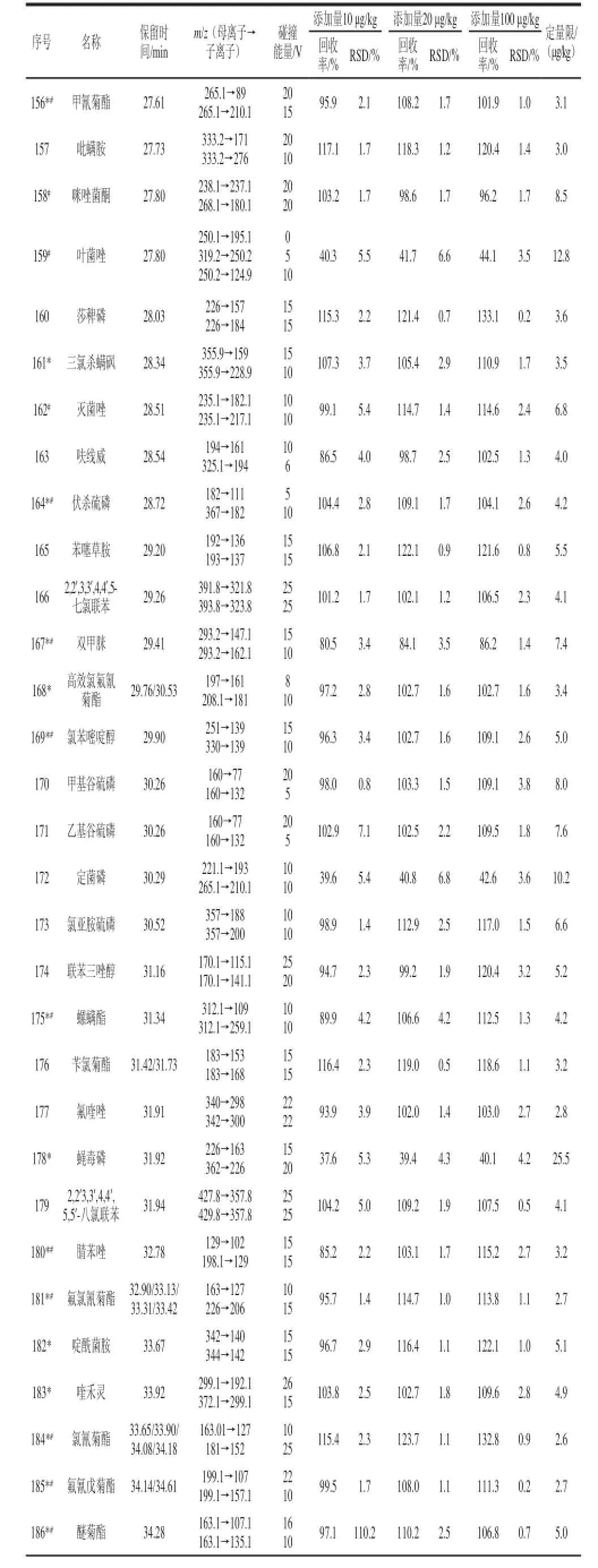
續表1

續表1
因PSA對脂肪酸、有機酸以及某些記性色素和糖類具有強親和性和高容量,應用DSPE法對葉菜類、瓜菜類、莖類、塊根類等幾類蔬菜樣品進行前處理后,能除去絕大多數雜質,可應用于大多數農藥殘留檢測,其中苯達松、氟螨噻、葉菌唑、久效威、百菌清、定菌磷、蠅毒磷、殺草敏、敵菌丹、滅菌磷、亞胺硫磷、吡氟酰草胺、氟吡草胺等農殘加標回收率低于50%,經查閱資料和反復實驗,敵菌丹在乙腈中,氟螨噻、蠅毒磷在二氯甲烷中,苯達松、葉菌唑、久效威、定菌磷、殺草敏、吡氟酰草胺、氟吡草胺等在丙酮中,百菌清、滅菌磷、亞胺硫磷等在甲苯中溶解度更大,用丙酮、甲苯、二氯甲烷等為提取溶劑再進行加標實驗,回收率均明顯提高。
2.6 實際樣品測定

圖3 陽性樣品的色譜圖及質譜圖Fig.3 TIC chromatogram and MS-MS spectrum for a positive sample
使用該方法對生菜、菜心、黃瓜、芹菜等50多個尚未上市銷售的樣品進行了檢測,結果在5 個樣品中檢出了毒死蜱、甲胺磷和氯氰菊酯3 種農藥,其含量均在15 μg/kg以下,毒死蜱和氯氰菊酯特征色譜圖及質譜圖見圖3。
3 結 論
本實驗建立了蔬菜中195 種農藥快速、靈敏、準確、高通量的DSPE-GC-MS-MS檢測方法。相對于傳統檢測方法具有簡便、快速、試劑消耗量小、選擇性好、靈敏度高、抗基質干擾能力強的優勢,滿足國內對于蔬菜中農藥殘留的測定要求,適用于蔬菜樣品中195 種農藥殘留量的快速篩查和定性確證檢測。
[1] ADOU K, BONTOYAN W R, SWEENEY P J. Multiresidue method for the analysis of pesticide residues in fruits and vegetables by accelerated solvent extraction and capillary gas chromatography[J]. Journal of Agricultural and Food Chemistry, 2001, 49(9): 4153-4160.
[2] 沈丹玉, 湯富彬, 袁新躍, 等. 基質固相分散-氣相色譜檢測菊花中有機磷農藥殘留[J]. 食品科學, 2012, 33(18): 216-219.
[3] 王素方, 張西安, 張東飛, 等. 氣相色譜法快速測定杜仲葉中有機氯菊酯類農藥殘留[J]. 食品科學, 2009, 30(18): 323-326.
[4] 李永新, 孫成均, 趙劍虹, 等. 高效液相色譜法同時測定蔬菜水果中的12種農藥殘留[J]. 色譜, 2006, 24(3): 251-255.
[5] 宋淑玲, 李重九, 馬曉東, 等. 蔬菜中殘留農藥的石墨化碳黑凈化和氣相色譜-質譜檢測方法[J]. 分析化學, 2008, 36(11): 1526-1530.
[6] 王明泰, 牟峻, 吳劍, 等. GC-MS法測定糧谷及油料中55 種有機磷農藥殘留量[J]. 分析試驗室, 2006, 25(11): 110-117.
[7] 曹殿潔, 黃信龍. 分散固相萃取-氣相色譜-質譜聯用法快速檢測蔬菜中18種農藥殘留[J]. 食品科學, 2013, 34(10): 219-222.
[8] 李巖, 鄭鋒, 王明林, 等. 液相色譜-串聯質譜法快速篩查測定濃縮果蔬汁中的156種農藥殘留[J]. 色譜, 2009, 27(2): 127-137.
[9] 王素利, 任麗萍, 劉聰云, 等. 分散固相萃取凈化液相色譜-質譜聯用快速檢測糙米中的多種殘留農藥[J]. 分析實驗室, 2009, 28(4): 38-42.
[10] 柳菡, 徐錦忠, 丁濤, 等. 蔬菜中26 種農藥殘留的高效液相色譜-串聯質譜法測定[J]. 分析測試學報, 2009, 28(2): 181-185.
[11] 李蓉, 盧俊文, 楊汝輝, 等. 氣相色譜-串聯質譜法測定水果中多種農藥殘留[J]. 中國衛生檢驗雜志, 2012(11): 2562-2565.
[12] 沈偉健, 余可垚, 桂茜雯, 等. 分散固相萃取-氣相色譜-串聯質譜法測定蔬菜中107種農藥的殘留量[J]. 色譜, 2009, 27(4): 391-400.
[13] 施家威, 李繼革, 王玉飛, 等. 分散固相萃取-氣相色譜-三重四極桿質譜分析蔬菜中112種農藥殘留[J]. 色譜, 2012, 30(6): 602-612.
[14] 王磊, 張麗杰, 呂偉, 等. 生物素-親和素放大酶聯免疫吸附法測定二乙基磷酸酯類有機磷農藥[J]. 分析化學, 2011, 39(3): 346-350.
[15] 劉浩, 周芳, 李月娥. 分散固相萃取法對果蔬中農藥殘留前處理的優化[J]. 環境科學與管理, 2008, 33(5): 137-139.
[16] 鄭文慧, 葉江雷, 王秀彬, 等. 分散固相萃取法在農藥殘留檢測中應用的進展[J]. 分析儀器, 2011(4): 23-27.
[17] 郝開拓, 孔祥虹, 杜寶中, 等. 分散固相萃取-氣相色譜-質譜法分析蕨菜-黑米中53種農藥殘留量[J]. 分析試驗室, 2011, 30(6): 69-74.
[18] 陳健航, 葉瑜霏, 程雪梅, 等. 分散固相萃取-氣相色譜-質譜聯用法檢測蔥-韭菜和姜中多種農藥殘留[J]. 質譜學報, 2011, 32(6): 341-349.
Simultaneous Determination of 195 Pesticide Residues in Vegetables Using Solid Phase Dispersive Extraction-Gas Chromatography-Tandem Mass Spectrometry
LI Rong1, LU Jun-wen1, YANG Fang1, ZHANG Peng-jie1, LI Yong-jun2, XIE Jian-jun3, CHU Da-ke4
(1. Zhongshan Entry-Exit Inspection and Quarantine Bureau, Zhongshan 528403, China; 2. Zhongshan Quality Supervision and Inspection Institute of Agricultural Products, Zhongshan 528400, China; 3. Guangdong Entry-Exit Inspection and Quarantine Bureau, Guangzhou 510623, China; 4. Guangdong Pharmaceutical University, Zhongshan 528400, China)
Objective: To develop a method for the determination of 195 pesticide residues in vegetables using gas chromatography with tandem mass spectrometry (GC-MS-MS) under a selected reaction monitoring (SRM) mode. Methods: The target compounds were extracted from samples with ethyl acetate and the extract was cleaned up by off-line solid phase dispersive extraction technique. The analytes were quantified by an internal standard method. Results: In the linear range (0.01–1.00 mg/L), the correlation coefficient for each pesticide was higher than 0.99. The average recoveries at three spiked levels (10, 20 and 100 μg/kg) varied from 37.6% to 136.7% with relative standard deviations (RSDs) between 0.2% and 15.3%. The limits of quantification (LOQ) (signal/noise ratio = 10) were 1.9–25.5 μg/kg. Conclusion: The method is rapid, sensitive, accurate and high throughput, without matrix interference and suitable for the analysis of 195 pesticide residues in vegetables.
solid phase dispersive extraction; gas chromatography-tandem mass spectrometry (GC-MS-MS); pesticide residues; vegetable
TS207.5
A
1002-6630(2014)24-0301-07
10.7506/spkx1002-6630-201424058
2014-04-01
中山市科技計劃—農業科技攻關計劃項目(20103A190);中山市科技發展專項(2013A3FC0251)
李蓉(1969—),女,高級工程師,本科,研究方向為食品安全檢測技術。E-mail:lir@zs.gdciq.gov.cn
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
