先天性肌營養不良1A型顱腦磁共振成像和基因檢測診斷分析:附兩例報道及文獻復習
2014-02-08 06:11:06張曉莉牛國輝杜開先徐發林賈天明
中國全科醫學 2014年24期
關鍵詞:基因突變
張曉莉,牛國輝,杜開先,徐發林,韓 瑞,賈天明
先天性肌營養不良(congenital muscular dystrophy,CMD)是一組異質性遺傳性神經肌肉病,該疾病的分型有十余種,多以不同的致病基因為主,其中的先天性肌營養不良1A型(merosin-deficient congenital muscular dystrophy,MDC1A)與編碼laminin-α2(又稱merosin)的LAMA2基因突變有關,又稱Merosin缺陷型,占總CMD的30%~40%,但該型在歐洲常見,我國報道很少,迄今國內共報道13例,僅3例有基因診斷[1-3]。現報道我院收治的2例MDC1A患兒,并結合國內報道的13例患兒做一總結,研究顱腦磁共振成像(MRI)和基因檢測診斷的意義。
1 資料與方法
1.1 病例資料 先證者1,女,8月齡,因下肢無力于2013-07-16就診。系第1胎第1產,足月順生,無窒息。4月齡時發現扶站下肢不支撐,未行治療,8月齡仍不能扶站,遂來我院就診。查體:智能、語言正常,豎頭穩,雙手抓物靈活,可獨坐,扶站下肢不支撐,膝腱反射消失,雙下肢肌張力低,肌力4級,肌容積正常,上肢肌力正常。既往未曾治療,家族史無特殊。查肌酸激酶(CK)2 500 U/L。肌電圖示肌源性損害。顱腦MRI示腦白質營養不良(見圖1)。診斷MDC1A。提取外周抗凝血4 ml,送康旭醫學檢驗所通過二代測序法進行基因檢測,并通過一代測序法進行驗證,LAMA2基因檢測發現復合雜合突變,一個是無義突變c.4048C>T(p.Arg1350*),一個是移碼突變c.457-458insT(p.Ile153fs),均為雜合突變(見圖2)。
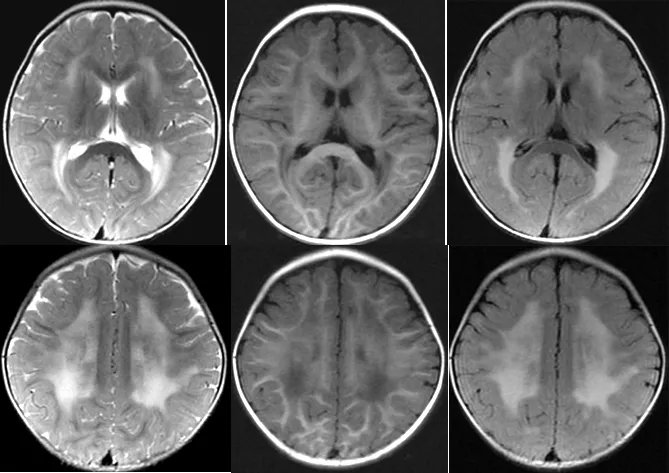
圖1 先證者1顱腦MRI示腦白質廣泛融合性脫髓鞘病變
Figure1 The brain MRI of proband 1 showing widely distributed fusion of white matter demyelination
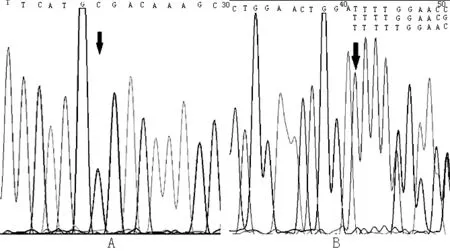
注:A圖為無義突變,編碼區第4048號堿基由C變為T,導致本該編碼精氨酸的1350號密碼子變為終止密碼子;B圖為移碼突變,在編碼區457號堿基和458號堿基之間插入了T,導致第153號密碼子后發生移碼變異
圖2 先證者1 LAMA2基因測序圖
Figure2 LAMA2-gene sequencing diagram of proband 1
先證者2,男,3歲4月,因發育落后,下肢無力,不會獨站,遂于2013-10-25就診。系第1胎第1產,足月剖宮產,出生體質量2 800 g,生后哭聲稍低,無窒息搶救史。運動發育史:9個月時翻身,1歲會坐,現不會獨站。查體:語言表達良好,智力正常,可獨坐,不能站立,上肢肌力4級,雙下肢肌力3級,雙膝反射及跟腱反射消失,雙足輕度內翻,無明顯肌肉萎縮,四肢肌張力低下,以下肢明顯。肌電圖提示:神經肌肉混合性損害,CK 1 041 U/L,顱腦MRI示雙側腦室前角旁、體旁及后角旁對稱性異常信號(見圖3)。診斷MDC1A。
1.2 文獻復習 通過萬方數據庫和中國知網中文數據庫以“肌營養不良”和“先天性”為檢索詞在題名或關鍵詞中檢索2013年12月以前發表的中文學術期刊,并在PubMed數據庫中查到中國人發表的先天性肌營養不良文獻1篇,通過閱讀全文,從中篩選出符合MDC1A的病例,共9 篇文獻,13例患兒,結合本院的2例患兒,共15例,做一綜合分析。
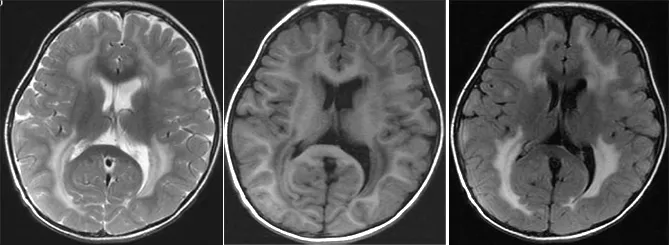
圖3 先證者2顱腦MRI示側腦室前角、體部、后角旁均可見融合性脫髓鞘病變
Figure3 The brain MRI of proband 2 showing the integration of demyelination in the front horn,the body and beside the posteior horn of lateral ventricle
2 結果
15例患兒中運動發育落后15例,CK升高14例,顱腦MRI彌漫性白質異常信號 14例,智力正常13例,腱反射消失13例(2例未描述),肌電圖檢查11例均顯示肌源性損害,并周圍神經損害2例,病理檢查9例均為肌營養不良改變;基因檢測4例,均發現LAMA2基因突變,其中2例為純合突變,2例為復合雜合突變(見表1)。
3 討論
MDC1A是最常見的CMD,歐美多見,我國罕見,目前報道共13例(其中臨床診斷2例,病理診斷8例,基因診斷3例),其確診目前主要依靠肌活檢,但此檢查方法多數醫院不能進行。本研究通過對15例患兒共同特點的分析發現,顱腦MRI在診斷上具有特異性,而進一步基因檢測可以確診并可以指導產前診斷。
3.1 臨床特點 回顧國內既往的病例報道和本文的2例共15例MDC1A患兒發現,所有病例在生后6個月內起病,生后早期即表現出明顯的肌張力低下,運動發育遲滯,以端坐延遲(13/15)、不能扶站就診者居多,最大獲得運動功能多為獨坐(13/15),僅2例獲得行走能力,但為肌病步態。體格檢查除發現肌張力低下、運動落后,均可見腱反射消失(13/13,2例未描述),此與中樞性疾病所導致的肌張力低下和發育落后不同。
血清學檢查見CK水平明顯升高,數倍至數十倍升高,但無臨床心肌受損的表現。本組患兒中CK最高達9 640 U/L。本病患兒肌電圖檢查主要呈肌源性損害(11/11),部分同時存在周圍神經損害(病例12和15),因為merosin蛋白除了存在于橫紋肌細胞的基底膜外,在腦血管基底膜、發育中的白質纖維束均有表達,還存在于施旺細胞的基底膜,因此周圍神經系統也可受累,表現為運動或感覺神經傳導速度減慢[10],本文先證者2肌電圖檢查即發現脛后、腓總神經運動傳導速度減慢,感覺傳導未見異常。肌肉活檢9例均呈肌營養不良改變,表現為肌纖維的壞死、再生,肌間纖維結締組織增加,還有炎性細胞浸潤,用特異性抗體行免疫組織化學染色顯示merosin染色陰性,提示merosin蛋白缺失。merosin蛋白是層黏連蛋白-2的3個亞單位之一,參與細胞間的識別、細胞分化、塑形和遷移等,merosin缺乏可造成細胞骨架與細胞外基質的連接破壞,導致肌纖維變性、壞死。
3.2 顱腦MRI改變 MDC1A患兒多無明顯的認知功能障礙(15例中僅2例智力稍差,余均正常),但常規行顱腦MRI檢查均可見腦白質營養不良改變(15例患兒中有1例未描述顱腦MRI改變,其余14例均有該特異性改變),與常見的腦白質營養不良均有嚴重的認知功能障礙不同。這些腦白質病變的特點均為雙側對稱,多為彌漫性白質異常,側腦室旁白質和皮質下均可見到。既往認為merosin陰性患兒僅有腦白質的病變,近年來也有小腦、腦干發育不良的報道[11]。本文2例患兒顱腦MRI均表現為側腦室前角、體部、后角旁廣泛融合性脫髓鞘改變,未見小腦和腦干的異常。此種特征性的改變有助于本病的診斷,其病理學機制尚不清楚。
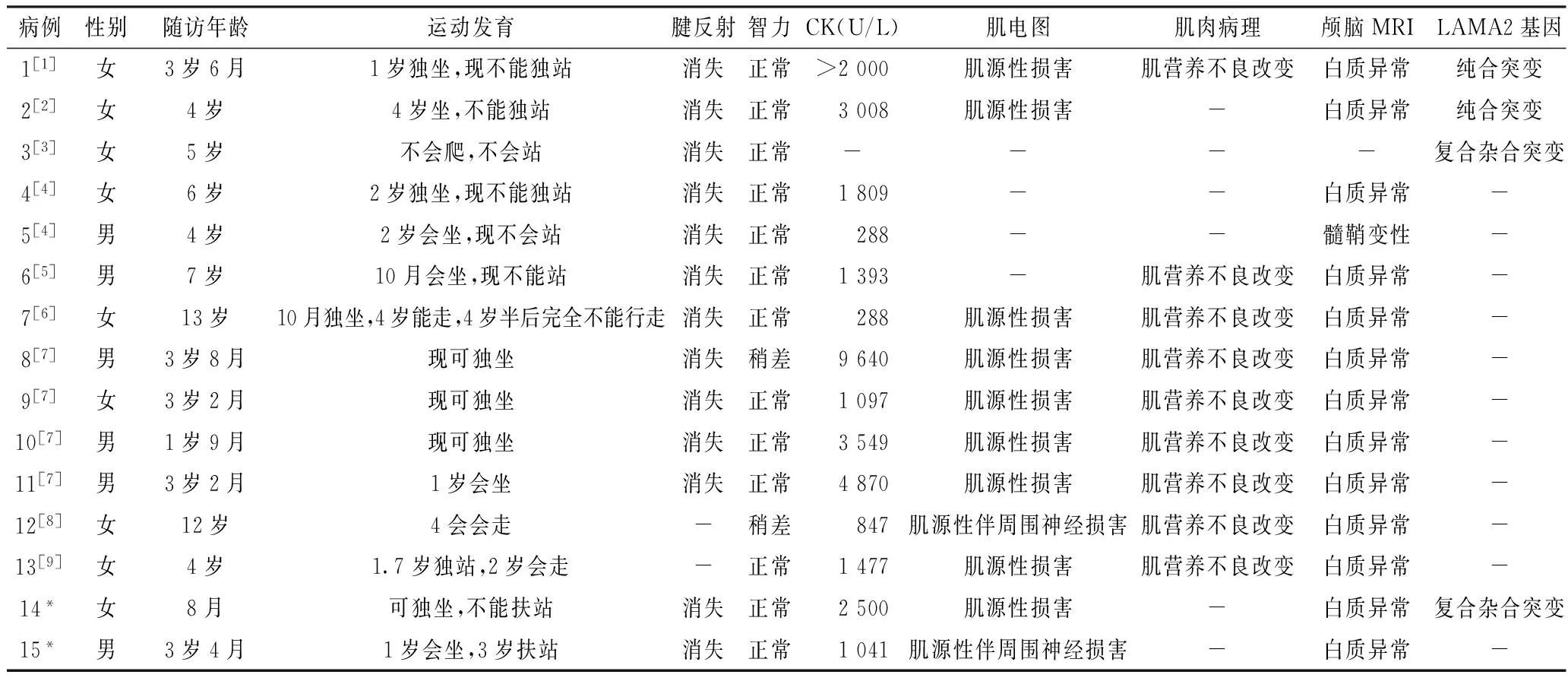
表1 15例MDC1A患兒的臨床資料
注:*為本文病例,-為無此項;CK=肌酸激酶
因此,本文認為對因肢體無力就診的患兒,若發現激酶譜明顯升高,肌電圖提示肌源性損傷改變,應常規進行顱腦MRI的檢查,若存在腦白質營養不良的改變,應高度考慮MDC1A。
3.3 基因改變 MDC1A由LAMA2基因突變所致,基因定位在6q22-23,含65個外顯子,突變覆蓋9 500 bp的編碼序列。突變類型包括無義、錯義、缺失、重復以及剪接位點的改變。近些年歐美對其基因突變的類型做了大量的研究。2008年Oliveira等[12]對來自于葡萄牙、西班牙、瑞士等國家的26例患兒進行基因分析,發現18種基因突變;2010年Geranmayeh等[13]對51例患兒進行基因分析,發現44種基因的突變類型。由于遺傳異質性的存在,同一種突變類型臨床表現可以相差甚遠[14],其不同突變類型與表型的關系尚需進一步研究。
本文先證者1進行LAMA2基因檢測,因LAMA2基因龐大,突變類型多樣,直接測序費用高,本研究采用二代測序法,結果在LAMA2基因上發現2個位點的雜合突變,一個為無義突變,致病性之前已有報道[13],編碼區第4048號堿基由C變為T,導致本該編碼精氨酸的1350號密碼子變為終止密碼子;第二個突變為移碼突變,在編碼區457號堿基和458號堿基之間插入了T,導致第153號密碼子后發生移碼變異,為國際上尚未報道的新發突變。本例發現的基因突變雖不是等位基因的兩個相同位點上發生的純合突變,但兩個雜合突變均發生在LAMA2基因,且均為致病性突變,可以導致發生MDC1A的表型。本例因故未能對先證者父母進行基因檢測,因先證者父母表型正常,MDC1A為常染色體隱性遺傳,推測此兩個突變分別來自父親、母親,即其父母分別各攜帶一個雜合突變而不致病,符合常染色體隱性遺傳的規律。這種遺傳方式既往曾有報道[3]。
15例患兒中有4例進行基因檢測,均發現LAMA2基因突變,其中2例為純合突變,2例為復合雜合突變,均符合常染色體隱性遺傳的規律。說明基因檢測對該病診斷具有確定意義。
該病目前無特效治療,臨床只能給予對癥處理,改善功能緩解病情進展。有關動物實驗的研究近年來較多,如應用層黏連蛋白111[15]、血管緊張素Ⅱ的1型受體拮抗劑[16]、整合素α7[17]等改善病變肌肉的病理結構和提高生存能力,希望不久的將來能用于臨床,改善患兒的生活質量。
MDC1A系罕見病,致死率、致殘率高,肌肉活檢可以用于先證者的診斷,但無法進行產前檢查,因此盡量明確先證者的基因類型,指導優生優育、進行產前診斷是預防該病患兒出生的惟一手段。
1 王碩,熊輝,羅靜,等.一個先天性肌營養不良1A型家系的臨床、分子病理及遺傳學研究[J].中華醫學遺傳學雜志,2010,27(1):13-16.
2 He Z,Luo X,Liang L,et al.Merosin-deficient congenital muscular dystrophy type 1A:A case report[J].Exp Ther Med,2013,6(5):1233-1236.
3 朱艷慧,喻長順,王曉春,等.層黏連蛋白α2缺失型先天性肌營養不良患兒一例LAMA2基因突變分析[J].中華臨床醫師雜志:電子版,2013,7(13):5871-5874.
4 吳元重,徐方元,吳綱烈.先天性肌營養不良-非福山型2例[J].安徽醫學,2000,21(1):63.
5 龍冬蘭,蔡少華,林茂增,等.Merosion 缺失型先天性肌營養不良一例[J].中華醫學遺傳學雜志,2005,22(4):469.
6 劉建國,沈定國.Merosin蛋白缺陷性先天性肌營養不良一例[J].中華神經科雜志,2006,39(4):263.
7 熊暉,姚生,袁云,等.先天性肌營養不良的診斷及層黏連蛋白表達的意義[J].中華兒科雜志,2006,44(12):918-923.
8 朱雯華,趙重波,林潔,等.先天性肌營養不良1A型的臨床表現和病理改變(附1例報道)[J].中國臨床神經科學,2008,16(5):504-508.
9 閆炳蒼,李建軍.先天性肌營養不良合并腦白質營養不良附1例報道[J].罕少疾病雜志,2010,17(1):50-51.
10 Quijano-Roy S,Renault F,Romero N,et al.EMG and nerve conduction studies in children with congenital muscular dystrophy[J].Muscle Nerve,2004,29(2):292-299.
11 劉茜瑋,肖江喜,謝晟,等.MRI對先天性肌營養不良的診斷價值[J].臨床放射學雜志,2010,29(4):506-509.
12 Oliveira J,Santos R,Soares-Silva I,et al. LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients[J].Clin Genet,2008,74(6):502-512.
13 Geranmayeh F,Clement E,Feng LH,et al.Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations[J].Neuromuscul Disord,2010,20(4):241-250.
14 Di Blasi C,Bellafiore E,Salih MA,et al.Variable disease severity in Saudi Arabian and Sudanese families with c.3924 + 2 T > C mutation of LAMA2[J].BMC Res Notes,2011,4:534.
15 Rooney JE,Knapp JR,Hodges BL,et al.Laminin-111 protein therapy reduces muscle pathology and improves viability of a mouse model of merosin-deficient congenital muscular dystrophy[J].Am J Pathol,2012,180(4):1593-1602.
16 Meinen S,Lin S,Ruegg MA.Angiotensin Ⅱ type 1 receptor antagonists alleviate muscle pathology in the mouse model for laminin-α2-deficient congenital muscular dystrophy(MDC1A)[J].Skelet Muscle,2012,2(1):18.
17 Doe JA,Wuebbles RD,Allred ET,et al.Transgenic overexpression of the α7 integrin reduces muscle pathology and improves viability in the dy(W) mouse model of merosin-deficient congenital muscular dystrophy type 1A[J].J Cell Sci,2011,124(Pt 13):2287-2297.
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
