1,3-二羥基丙酮的合成與應用研究進展
2014-01-14 09:04:40裴承強陳建華
化學與生物工程 2014年8期
關鍵詞:效率
裴承強,陳建華
(中國藥科大學生命科學與技術學院分子生物學教研室,江蘇 南京210009)
1,3-二羥基丙酮(1,3-dihydroxyacetone,DHA)是一種最簡單的酮糖,具有3個活性基團,可參與多種反應,廣泛應用于化工、醫藥、化妝品和食品領域[1]。DHA的合成方法主要包括化學合成法和微生物合成法。由于化學合成法存在轉化效率低、選擇性氧化效率低、污染環境等問題,因此,研究者更傾向于環境友好、轉化效率高的微生物合成法,但微生物合成法也存在底物與產物抑制的問題。作者在此就DHA的兩種主要合成方法的研究進展進行了綜述,并對其在化工與醫藥領域的最新應用進展進行了簡單介紹。
1 DHA的合成
1.1 化學合成法
化學合成法包括甲醛縮合法和甘油氧化法,目前研究較多的是甘油氧化法。甘油氧化法采用重金屬作催化劑,存在轉化效率低、選擇性氧化效率低、污染環境等問題。
Hirasawa等[2]介紹了一種Pd-Ag/C作催化劑的路徑,其反應機制(圖1)為:(1)底物的末端羥基被吸附在Ag表面;(2)被吸附在Pd表面的活性氧攻擊破壞第二位上的C-H鍵,伴隨著第二位上O-H鍵的斷裂,羰基產物形成;(3)金屬表面的羥基被質子化并以水的形式離開,催化劑恢復初始金屬狀態。Pd單獨催化表現出的是低選擇性,Ag單獨催化表現出的是低活性,而Pd-Ag催化表現了高選擇性和高活性。Pd-Ag/C系統雖能在很長的反應時間內保證高選擇性(85%)氧化生成DHA,但存在酸中毒的問題,即反應會在生成大量酸性副產物甘油酸時,被Pd強烈吸附而封鎖活性氧的結合位點,致使反應終止。
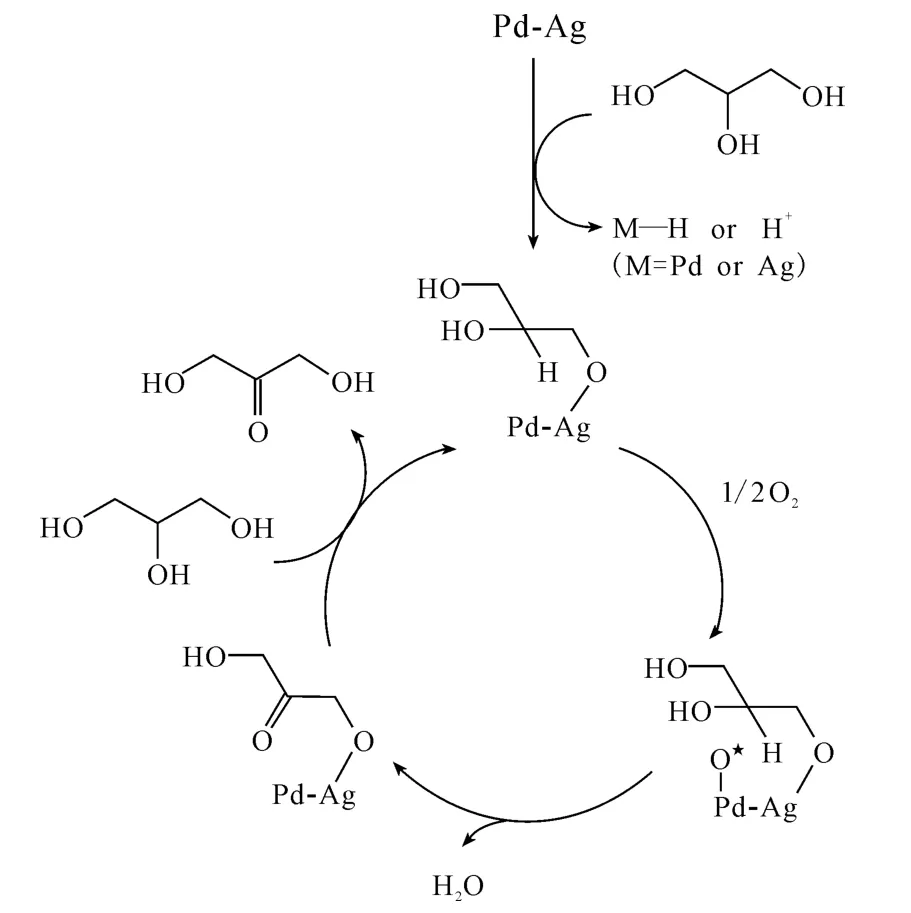
圖1 Pd-Ag/C作催化劑的甘油氧化法機制Fig.1 Mechanism of glycerol oxidation method using Pd-Ag/C as catalyst
Rodrigues等[3-4]設計了一種方法使得DHA的生產速率達到5.4g·L-1·h-1,是微生物氧化甘油(約1.8g·L-1·h-1)的3倍。該法以Au為催化劑、以多壁碳納米管(MWCNTs)為載體,DHA收率達到60%,而以活性炭為載體時的DHA收率僅為18%。
Wang等[5]通過甘油間接氧化法合成DHA,主要是通過磁性的聚苯乙烯納米球固定催化劑2,2,6,6-四甲基哌啶氧化物(TEMPO)完成,該法適用于多種醇的氧化,其機制如圖2所示。

圖2 HPD經過氧化合成DHAFig.2 Synthesis of DHA by HPD oxidation
HPD(1,3-acetalized glycerol)由甘油和苯甲醛通過縮醛反應制得。由于甘油的三羥基結構,如何控制氧化羥基的位置成為難題;同時重金屬的直接氧化法還存在產物的過氧化問題,導致DHA收率不高,也給分離純化帶來困難。該法將末端羥基保護,成功克服了選擇性氧化的難題;同時將昂貴的TEMPO固定化,不僅可以重復使用,而且解決了TEMPO在分離純化和結晶時帶來的難題,DHA轉化效率達到95.3%,結晶純度>99%,收率為78.1%。
Zhang等[6]通過一種以納米粒子為支撐的多酶原位輔因子再生系統,構建出一條以NAD(H)為輔因子、甘油脫氫酶(GDH)和木糖還原酶(XR)參與的新途徑,其機制如圖3所示。

圖3 GDH/XR多酶系統模型Fig.3 The model of GDH/XR multi-enzyme system
相較于原生系統,由納米粒子作為支撐的多酶系統提高了酶的穩定性和可重復利用性,甘油脫氫酶可被重復利用多次,DHA總收率達到160g·(gGDH)-1。該途徑提出了一種不同于天然生物代謝途徑的可高效獲得高附加值化合物和材料的新策略。
1.2 微生物合成法
相較于化學合成法,微生物合成法有其獨特的優勢,如對環境無污染、對二位羥基選擇性氧化能力高、轉化效率高、易操作、反應條件溫和等,但也存在一些問題,如菌體對底物濃度的耐受能力有限、產物濃度對代謝酶的抑制以及對細胞的損傷、發酵周期長等。菌株改造可以彌補底物與產物對菌株的抑制、酶活不高、發酵周期長等不足。常用的菌株改造手段包括誘變和基因工程改造。
1.2.1 誘變
誘變包括輻射誘變、化學誘變等。Ma等[7]采用He-Ne輻射對氧化葡萄糖酸桿菌進行誘變,確定21 mW、21min為最優誘變條件,獲得的突變株GM51的甘油脫氫酶活性比原始菌株提高了75%,轉化效率達到91.5%,生產速率提高77.6%,發酵周期縮短了16h。Hu等[8]采用離子注入法誘變氧化葡萄糖酸桿菌之后通過響應面優化培養條件,選取最優條件60×(2.6×1013)ions·cm-2、能量為10keV進行誘變,獲得突變株G.oxydans ZJB09113,響應面優化后,DHA產量提高196.3%。Hu等[9]還采用紫外誘變獲得一株DHA高產突變株G.oxydans ZJB11001,在此基礎上控制溶氧濃度獲得產量為(209.6±6.8)g·L-1的DHA。
1.2.2 基因工程改造
主要從三個方面進行基因工程改造:(1)敲除adhA基 因:Habe等[10-11]研 究 了 敲 除adhA基 因(ΔadhA)對轉化甘油生成甘油酸的必要性,ΔadhA菌株能提高DHA產量,并能耐受高濃度甘油,對照菌株在220g·L-1甘油濃度下基本不生長,而ΔadhA菌株則可產生125g·L-1的DHA。(2)過表達甘油脫氫酶基因sldAB:Gatgens等[12]通過過表達sldAB基因提高了DHA產量,對照菌株在550mmol·L-1甘油濃度下產生200~280mmol·L-1DHA,而突變株則可產生350mmol·L-1DHA。(3)對同一菌株同時敲除adhA基因和過表達sldAB基因:Li等[13]通過敲除adhA基因和過表達sldAB基因提高了DHA產量并縮短了發酵周期,突變株轉化100g·L-1甘油生成96g·L-1DHA,生產速率從對照菌株的1.0g·(gCDW)-1·h-1提高到2.4g·(gCDW)-1·h-1,并且當耐受140g·L-1甘油時,突變株能產生134g·L-1DHA。
Nguyen等[14]在釀酒酵母菌中利用糖類代謝以甘油為中間代謝物生產DHA,將2個磷酸化DHA的DAK1/DAK2酶基因敲除,阻斷了DHA的去路,有利于產物的累積,但轉化效率不是很高。
1.3 其它方法
優化發酵工藝也可獲得高產量的DHA。Hekmat等[15]用一種涂有多孔硅基質的Ralu-rings載體對細胞進行固定化,進行半連續重復分批補料發酵,顯著提高了空間-時間產率。Hu等[16]用氣升式發酵罐補料生產DHA,在保證轉化效率達到(89.8±2.4)%的基礎上,生產速率可達到2.17g·L-1·h-1。Hu等[17]在分批補料發酵生產DHA的過程中,通過設定溶氧參數調節甘油的流加,同時根據菌體生長和發酵時對pH值的不同需求,對發酵液pH值進行調節,有效地解決了底物抑制、高濃度氧損傷細胞、低濃度氧不利于甘油轉化的問題。Liu等[18]分離篩選出一株新菌株,以生物柴油副產品粗制的甘油為底物可以獲得轉化效率為90.5%、生產速率為2.6g·L-1·h-1的DHA。
2 DHA的應用
2.1 化工領域
DHA除可參與曼尼西反應、美拉德反應外,還可參與高碳糖化合物的合成以及乳酸衍生物合成。庚糖類、辛糖類、壬糖類等高碳糖化合物在生物細胞中扮演著重要角色,Cieplak等[19]通過封閉羥基的DHA與D-阿拉伯糖合成了高碳糖。
乳酸衍生物是一種新型的綠色溶劑,可以溶解醋酸纖維素、硝酸纖維素、油類、染料類、涂劑等。乳酸乙酯可替代工業上傳統有毒的鹵代有機介質[20],乳酸丙酯可作為藥物和農藥合成的手性中間體。DHA經脫水生成丙酮醛(methylglyoxal,MGO),而后與相應醇可生成該類衍生物[21]。Mylin等[22]將DHA在乙醇溶液中經過兩性氧化劑ZrO2-TiO2催化,獲得了90%的乳酸乙酯。
2.2 醫藥領域
在Manuka蜂蜜貯藏過程中,其富含的DHA會轉化為MGO,而富含DHA和MGO的Manuka蜂蜜可作為廣譜藥物抑制微生物(包含多重耐藥菌),同時可作為傷口敷劑治愈創傷[23]。
通過抑制α糖苷酶活性治療Ⅱ型糖尿病已成為醫藥領域的研究熱點。多羥基哌啶化合物(含氮多羥基糖)具有顯著的糖苷酶抑制活性,糖苷酶抑制劑Fagomine是一種亞氨基糖,通過DHA與N-Cbz-3-aminopropanal的羥醛縮合反應可以合成其前體,6-磷酸果糖醛縮酶(FSA)的2種突變體A129S和A129S/165G可以催化該反應(圖4)[24-25]。

圖4 DHA與N-Cbz-3-aminopropanal的羥醛縮合反應Fig.4 Aldol reaction of DHA and N-Cbz-3-aminopropanal
DHA能夠抑制敲除DHA激酶基因酵母菌的生長,能夠自由通過瘧原蟲屬非水特異性通道,而瘧原蟲屬本身不含DHA激酶基因,因而同樣可以產生上述效應。DHA可被人紅細胞作為能源利用,由于瘧原蟲缺乏DHA激酶,因而不能利用,同時DHA又能抑制瘧原蟲磷酸甘油醛脫氫酶(PfGAPDH),阻斷其能量來源,妨礙其增殖,可以被設計成一種以GAPDH為靶點的抗瘧疾藥物[26]。DHA還能夠使錐形蟲的細胞周期在G2/M期停滯,影響其增殖。
磷化氫能夠抑制大腦、心臟、肝臟的細胞色素C氧化酶的活性,而DHA可以解除該抑制作用[27-28],不僅如此,DHA還可解氰化物毒性。此外,DHA可用作食品添加劑、防腐劑等,還可用來治療白癜風,提高瘦肉率,達到減肥效果。
3 展望
DHA作為一種重要的化工醫藥中間體,應用廣泛,潛力巨大。化學合成法在甘油羥基的選擇性氧化、轉化效率、生產速率等方面取得了突破性進展,菌株改造也獲得很大突破。為提高甘油利用率和DHA轉化效率,今后應針對DHA合成中所存在的問題,優化合成方法和發酵工藝。
[1]MISHRA R,JAIN S R,KUMAR A.Microbial production of dihydroxyacetone[J].Biotechnol Adv,2008,26(4):293-303.
[2]HIRASAWA S,WATANABE H,KIZUKA T,et al.Performance,structure and mechanism of Pd-Ag alloy catalyst for selective oxidation of glycerol to dihydroxyacetone[J].Journal of Catalysis,2013,300:205-216.
[3]RODRIGUES E G,CARABINEIRO S A C,DELGADO J J,et al.Gold supported on carbon nanotubes for the selective oxidation of glycerol[J].Journal of Catalysis,2012,285(1):83-91.
[4]RODRIGUES E G,PEREIRA M F R,DELGADO J J,et al.Enhancement of the selectivity to dihydroxyacetone in glycerol oxidation using gold nanoparticles supported on carbon nanotubes[J].Catalysis Communications,2011,16(1):64-69.
[5]WANG J L,ZHANG M,ZHENG Z,et al.The indirect conversion of glycerol into 1,3-dihydroxyacetone over magnetic polystyrene nanosphere immobilized TEMPO catalyst[J].Chemical Engineering Journal,2013,229(1):234-238.
[6]ZHANG Y,GAO F,ZHANG S P,et al.Simultaneous production of 1,3-dihydroxyacetone and xylitol from glycerol and xylose using a nanoparticle-supported multi-enzyme system with in situ cofactor regeneration[J].Bioresource Technology,2011,102(2):1837-1843.
[7]MA L J,LU W Y,XIA Z D,et al.Enhancement of dihydroxyacetone production by a mutant of Gluconobacter oxydans[J].Biochemical Engineering Journal,2010,49(1):61-67.
[8]HU Z C,LIU Z Q,XU J M,et al.Improvement of 1,3-dihydroxyacetone production from Gluconobacter oxydans by ion beam implantation[J].Preparative Biochemistry & Biotechnology,2012,42(1):15-28.
[9]HU Z C,ZHENG Y G.Enhancement of 1,3-dihydroxyacetone production by a UV-induced mutant of Gluconobacter oxydans with DO control strategy[J].Appl Biochem Biotechnol,2011,165(5-6):1152-1160.
[10]HABE H,FUKUOKA T,MORITA T,et al.Disruption of the membrane-bound alcohol dehydrogenase-encoding gene improved glycerol use and dihydroxyacetone productivity in Gluconobacter oxydans[J].Biosci Biotechnol Biochem,2010,74(7):1391-1395.
[11]HABE H,SATO S,FUKUOKA T,et al.Membrane-bound alcohol dehydrogenase is essential for glyceric acid production in Acetobacter tropicalis[J].Journal of Oleo Science,2011,60(9):489-494.
[12]GATGENS C,DEGNER U,BRINGER-MEYER S,et al.Biotransformation of glycerol to dihydroxyacetone by recombinant Gluconobacter oxydans DSM2343[J].Appl Microbiol Biotechnol,2007,76(3):553-559.
[13]LI M H,WU J,LIU X,et al.Enhanced production of dihydroxyacetone from glycerol by overexpression of glycerol dehydrogenase in an alcohol dehydrogenase-deficient mutant of Gluconobacter oxydans[J].Bioresource Technology,2010,101(21):8294-8299.
[14]NGUYEN H T,NEVOIGT E.Engineering of Saccharomyces cerevisiae for the production of dihydroxyacetone(DHA)from sugars:A proof of concept[J].Metabolic Engineering,2009,11(6):335-346.
[15]HEKMAT D,BAUER R,NEFF V.Optimization of the microbial synthesis of dihydroxyacetone in a semi-continuous repeatedfed-batch process by in situ immobilization of Gluconobacter oxydans[J].Process Biochemistry,2007,42(1):71-76.
[16]HU Z C,ZHENG Y G,SHEN Y C.Use of glycerol for producing 1,3-dihydroxyacetone by Gluconobacter oxydans in an airlift bioreactor[J].Bioresource Technology,2011,102(14):7177-7182.
[17]HU Z C,ZHENG Y G,SHEN Y C.Dissolved-oxygen-stat fedbatch fermentation of 1,3-dihydroxyacetone from glycerol by Gluconobacter oxydans ZJB09112[J].Biotechnology and Bioprocess Engineering,2010,15(4):651-656.
[18]LIU Y P,SUN Y,TAN C,et al.Efficient production of dihydroxyacetone from biodiesel-derived crude glycerol by newly isolated Gluconobacter frateurii[J].Bioresource Technology,2013,142:384-389.
[19]CIEPLAK M,CEBORSKA M,CMOCH P,et al.Synthesis of higher carbon sugars from dihydroxyacetone and D-arabinose:An organocatalytic approach[J].Tetrahedron:Asymmetry,2012,23(15-16):1213-1217.
[20]PEREIRA C S M,SILVA V M T M,RODRIGUESA A E.Ethyl lactate as a solvent:Properties,applications and production processes—A review[J].Green Chemistry,2011,13:2658-2671.
[21]DAPSENS P Y,KUSEMA B T,MONDELLI C,et al.Galliummodified zeolites for the selective conversion of bio-based dihydroxyacetone into C1-C4alkyl lactates[J/OL].Journal of Molecular Catalysis A:Chemical,http://dx.doi.org/10.1016/j.molcata.2013.09.032.
[22]MYLIN A M,LEVYTSKA S I,SHARANDA M E,et al.Selective conversion of dihydroxyacetone-ethanol mixture into ethyl lactate over amphoteric ZrO2-TiO2catalyst[J].Catalysis Communications,2014,47:36-39.
[23]ATROTT J,HABERLAU S,HENLE T.Studies on the formation of methylglyoxal from dihydroxyacetone in Manuka(Leptospermum scoparium)honey[J].Carbohydrate Research,2012,361:7-11.
[24]SUDAR M,FINDRIK Z,VASIC-RACKI D,et al.Aldol addition of dihydroxyacetone to N-Cbz-3-aminopropanal catalyzed by two aldolases variants in microreactors[J].Enzyme and Microbial Technology,2013,53(1):38-45.
[25]GUTIERREZ M,PARELLA T,JOGLAR J,et al.Structureguided redesign of D-fructose-6-phosphate aldolase fromE.coli:Remarkable activity and selectivity towards acceptor substrates by two-point mutation[J].Chemical Communications,2011,47(20):5762-5764.
[26]PAVLOVIC-DJURANOVIC S,KUN J F,SCHULTZ J E,et al.Dihydroxyacetone and methylglyoxal as permeants of the Plasmodiumaquaglyceroporin inhibit parasite proliferation[J].Biochimica et Biophysica Acta,2006,1758(8):1012-1017.
[27]NIKNAHAD H,GHELICHKHANI E.Antagonism of cyanide poisoning by dihydroxyacetone[J].Toxicology Letters,2002,132(2):95-100.
[28]NIKNAHAD H,HASHEMI A,JAMSHIDZADEN A.Antidotal effect of dihydroxyacetone against phosphine poisoning in vivoin mice[J].Toxicology Letters,2012,211:S169-S170.
猜你喜歡
瘋狂英語·初中天地(2021年5期)2021-07-21 02:24:28
甘肅教育(2020年14期)2020-09-11 07:57:42
中學生數理化(高中版.高考數學)(2020年5期)2020-06-02 09:19:08
商周刊(2017年9期)2017-08-22 02:57:49
遼寧經濟(2017年6期)2017-07-12 09:27:16
中國衛生(2016年9期)2016-11-12 13:27:54
時代英語·高二(2015年1期)2015-03-16 00:08:11
中國洗滌用品工業(2015年7期)2015-02-28 19:02:38
電子設計工程(2015年12期)2015-02-27 12:06:10
中國衛生(2014年11期)2014-11-12 13:11:32
