典型單環和雙環芳烴加氫熱力學分析
2013-12-23 03:57:16侯朝鵬李永丹夏國富李明豐
石油化工 2013年6期
關鍵詞:催化劑
侯朝鵬,李永丹,夏國富,李明豐
(1. 中國石化 石油化工科學研究院,北京 100083;2. 天津大學 化工學院,天津 300072)
柴油中芳烴含量高不僅會降低油品的質量和十六烷值,還會增加柴油燃燒廢氣中的顆粒排放物,因而,芳烴加氫受到廣泛關注[1-3]。油品中的芳烴按芳環的數量主要分為4類:單環芳烴、雙環芳烴、三環芳烴和多環芳烴。在實驗研究中,通常選取苯及其同系物作為單環芳烴的模型反應物,萘及其同系物作為雙環芳烴的模型反應物。文獻[4-5]曾報道了幾種芳烴及其加氫產物的熱力學數據。Poling等[5-7]為解決芳烴熱力學數據缺乏的問題,將基團貢獻法用于估算某些芳烴及其加氫產物的標堆焓、熵和理想氣體熱容。Frye等[8-9]采用氣相體系測定了不同溫度和壓力下幾種芳烴/氫氣混合物的平衡組成,所選用的芳烴有聯苯、茚、萘、菲、二氫苊和芴。Jaffe[10]利用文獻[4]中的數據總結了烴類加氫放熱的規律,他認為不同種類烴的C—C鍵斷裂所釋放的能量不同,文獻[11]所報道的對苯加氫熱力學的計算結果與其一致。對含有支鏈的苯系芳烴,平衡常數隨側鏈數和每一個側鏈上碳原子數的增加而減小;萘系芳烴與苯系芳烴類似[12-14]。一般來說,含一個以上環的芳烴,其加氫是一個一個芳環逐步進行的。在傳統加氫條件下,第一個芳環的加氫平衡常數一般較高[12,15-16]。上述文獻均沒有考慮實際的反應過程和復雜的實驗條件。
對于實際的生產過程,可選擇幾種典型芳烴的加氫熱力學加以考慮,以尋找熱力學上合適的反應條件和操作區域,再綜合選擇它們的合適操作區域的公共部分,使不同的芳烴處于熱力學可行的加氫條件范圍之內。在實驗研究中,一般會針對反應過程選擇不同的模型反應物,而各種典型芳烴的加氫熱力學平衡計算會為芳烴加氫反應直接提供相關的熱力學允許的操作條件。
本工作利用熱力學數據庫軟件HSC-Chemistry 4.0對幾種典型的芳烴加氫反應熱力學平衡進行討論,充分考慮了反應條件的變化對芳烴加氫轉化率的影響,直接提供了熱力學平衡允許的操作條件。
1 熱力學計算依據及數據基礎
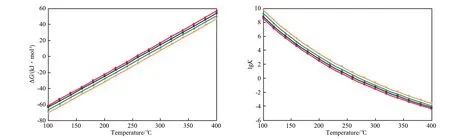
圖1 幾種單環芳烴加氫飽和反應的ΔG和lgK隨溫度的變化Fig.1 ΔG and lgK vs temperature in the saturate hydrogenation of several monocyclic aromatics.
利用商用軟件HSC-Chemistry4.0(Outokumpu公司產品)進行熱力學計算,各種物質的熱力學數據由軟件的數據庫引出。所有氣體均被認為是理想氣體;不特別指明時,所有反應以每mol主反應物為基準,反應熱的單位為kJ/mol;假定所有反應體系均為封閉體系,不特別指明時,壓力指絕對壓力,計算所得結果均為達到化學平衡時的組成,轉化率為平衡轉化率。苯加氫生成環己烷較為簡單,因此本工作采用苯的平衡轉化率對該過程進行描述;而萘的加氫產物較為復雜,為了表達方便,采用含氫基的平衡組成對該過程進行描述。
2 計算結果
2.1 單環芳烴加氫反應
一般常用的單環芳烴模型反應物有苯、甲苯、乙苯和二甲苯,以及它們的同分異構體和同系物。幾種單環芳烴加氫飽和反應的反應自由能變(ΔG)和lgK(K為反應平衡常數)隨溫度的變化見圖1。從圖1可見,這些反應的共同特點是:在100~400 ℃內,ΔG隨反應溫度的升高近似線性增大,K隨反應溫度的升高而降低。反應在低于250~300 ℃時為自發過程,反應溫度越低,ΔG越小,反應越傾向于自發進行。從圖1還可見,在相同反應溫度下,4種芳烴加氫反應的ΔG隨烷基側鏈上碳原子數的增加而增大,K隨烷基側鏈上碳原子數的增加而減小。這與文獻[12-14]報道的結果一致。
由于這些單環芳烴的熱力學反應過程及數據的規律性較為相似,因此,以苯為例進行計算,其他單環芳烴的計算結果與其類似。苯不僅是一種重要的化工原料,且在研究中經常作為單環芳烴加氫過程的模型反應物[17-27]。氣相苯的完全加氫產物是環己烷,該反應是體積縮小反應,增加系統壓力有利于苯加氫轉化率的提高;且該反應為強放熱反應,苯平衡轉化率隨溫度的升高而降低。影響苯平衡轉化率的因素較為復雜,本工作主要計算溫度、壓力和氫氣與苯的摩爾比(氫烴比)對苯平衡轉化率的影響。
溫度和氫烴比對苯加氫生成環己烷的平衡轉化率的影響見圖2。由圖2可看出,在各氫烴比下,平衡轉化率隨溫度的升高而降低。在氫烴比小于3時,由于每一個苯分子加氫生成環己烷要消耗3個分子的氫氣,如果氫氣量不足,會使苯轉化不完全;當氫烴比大于3時,氫氣過量,加氫后還有過量的氫氣存在,此時苯轉化率較高,且隨氫烴比的增加,平衡轉化率大于99.0%和99.9%的溫度范圍(等高線內側的兩條線內的區域,等高線是響應面在x-y平面上的投影)越來越大。
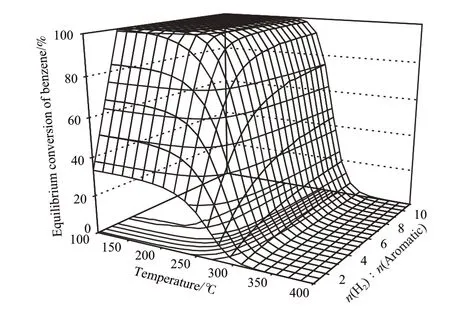
圖2 溫度和氫烴比對苯加氫生成環己烷的平衡轉化率的影響Fig.2 Effects of temperature and n(H2)∶n(aromatic) on the equilibrium conversion of benzene in its hydrogenation to cyclohexane.
在實際反應過程中,苯加氫反應一般在較高的氫烴比下操作。溫度和壓力對苯加氫生成環己烷的平衡轉化率的影響見圖3。
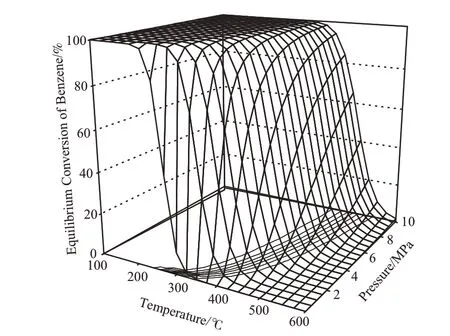
圖3 溫度和壓力對苯加氫生成環己烷的平衡轉化率的影響Fig.3 Effects of temperature and pressure on the equilibrium conversion of benzene in its hydrogenation to cyclohexane.
由圖3可看出,壓力為0.1 MPa時,在200 ℃以下,苯的平衡轉化率很高,在99.9%以上。隨溫度的升高,由于熱力學平衡的影響,苯的平衡轉化率逐漸降低;溫度高于350 ℃時,苯的平衡轉化率已很低。在每個溫度點,苯的平衡轉化率隨壓力的增加而增大,且隨壓力的增加,平衡轉化率超過99.9%和99.0%的溫度范圍越來越大,如在5.0 MPa時,苯的平衡轉化率超過99.9%的溫度范圍比0.1 MPa時升高了約100 ℃。
圖4是常規工業操作壓力(5.0 MPa)下,溫度和氫烴比對苯加氫生成環己烷的平衡轉化率的影響。由圖4可看出,在氫烴比低于3時,由于氫氣不足,平衡轉化率較低;且隨反應溫度的升高,平衡轉化率明顯降低,這與圖1中ΔG隨溫度的升高而增加,K隨溫度的升高而降低的趨勢一致。
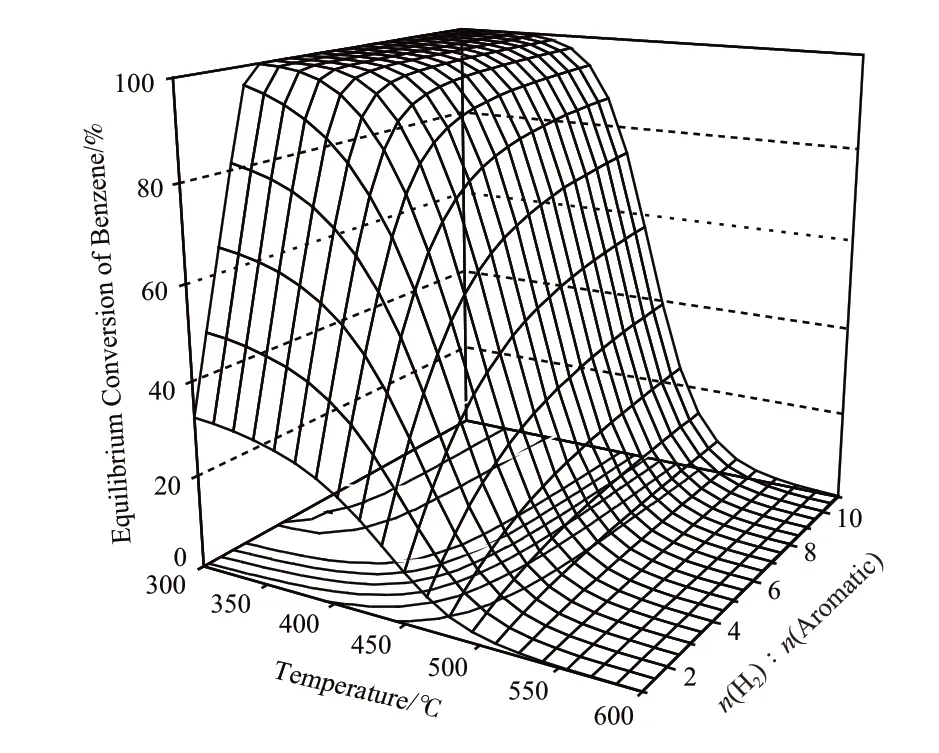
圖4 溫度和氫烴比對苯加氫生成環己烷的平衡轉化率的影響Fig.4 Effects of temperature and n(H2)∶n(aromatic) on the equilibrium conversion of benzene in its hydrogenation to cyclohexane.
從以上計算結果可知,溫度、氫烴比和壓力是影響苯平衡轉化率的重要因素。在選定反應溫度時,提高反應系統的壓力和氫烴比,都可使苯加氫生成環己烷的平衡轉化率增大。
在以苯為模型反應物的芳烴加氫系統中,由于使用的催化劑不同,反應的活性溫度范圍差異很大。不同壓力下Ni催化劑催化苯加氫反應的轉化率與溫度的關系見圖5。由圖5可見,若無Ni催化劑,苯加氫轉化率很低;采用Ni催化劑后,苯加氫轉化率明顯增加,且增加壓力對提高苯加氫轉化率有利[24]。
因此,為了尋找苯加氫合適的操作條件,必須充分綜合考慮催化劑、溫度、壓力和氫烴比等因素。圖2~4提供了苯加氫反應操作條件范圍的具體數值,這些數據為文獻[16]提供了有益的補充。由于圖1中的幾種主要單環芳烴模型物加氫飽和反應的ΔG和lgK隨溫度的變化情況一致,由此可以推測這幾種單環芳烴加氫反應的實驗數據也會與苯加氫反應的實驗數據呈相似的規律。
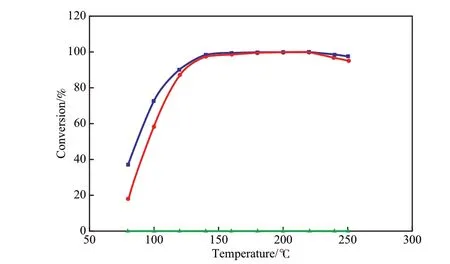
圖5 不同壓力下Ni催化劑催化苯加氫反應的轉化率與溫度的關系Fig.5 Conversion of benzene vs temperature under different pressure on Ni catalyst.
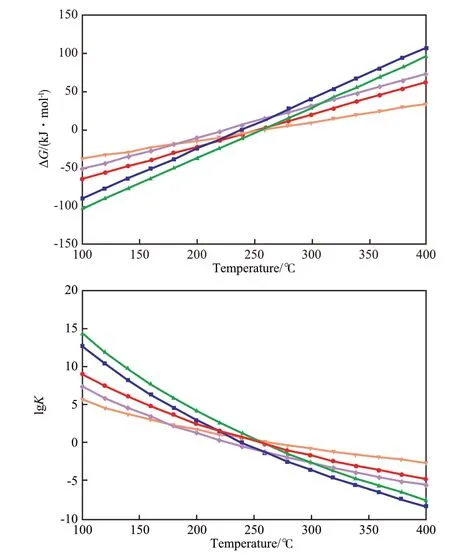
圖6 ΔG和lgK隨溫度的變化曲線Fig.6 ΔG and lgK vs temperature.
2.2 雙環芳烴加氫反應
雙環芳烴模型反應物的代表主要為萘。氣相萘加氫過程較復雜,一般有兩種穩定的加氫產物四氫萘和十氫萘,其中十氫萘有兩種同分異構體:順式十氫萘和反式十氫萘。主要反應的方程式如下:
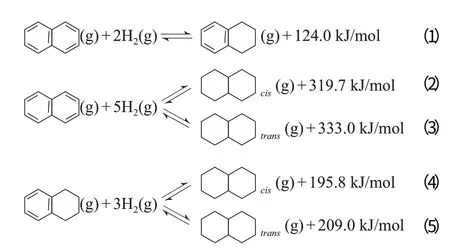
許多研究者[28-40]選擇萘作為芳烴加氫過程的模型反應物,也有研究者[41-50]選擇四氫萘。不同反應的ΔG和lgK隨溫度的變化見圖6。從圖6可見,這些反應的共同特點是:在100~400 ℃內,ΔG隨反應溫度的升高近似線性單調升高,K隨反應溫度的升高單調降低;在約250 ℃以下,ΔG為負值,說明這些反應在低于250 ℃時為自發過程,且反應溫度越低,ΔG越小,反應越傾向于自發進行。從圖6還可見,不同反應的ΔG隨溫度變化的趨勢有差別,其中萘加氫生成四氫萘的ΔG隨溫度的變化幅度最小。
萘體系加氫為體積縮小的反應,增加系統壓力有利于萘加氫轉化率的提高;提高氫氣分壓也有利于萘的轉化,且反應為強放熱反應,萘體系的平衡轉化率隨溫度的升高而降低。考慮到通常的反應狀況,選擇在氫氣過量下計算萘加氫過程的熱力學平衡組成。溫度和壓力對萘體系加氫產物分布的影響見圖7。由圖7可見,對應于每個系統壓力,反應過程中的組分都很復雜,一般是氫氣、萘、四氫萘和十氫萘的混合物。從圖7a可見,在該體系中,低溫下萘可以完全加氫轉化,但在不同壓力下,萘完全轉化的溫度不同,隨壓力的增加,萘完全加氫轉化的溫度范圍越來越寬。在高溫下,對應于每個壓力,萘的平衡轉化率隨溫度的增加而降低。而隨壓力的升高,對應較低萘平衡組成的溫度區逐漸變寬,說明系統壓力的升高有利于萘加氫,如在0.1 MPa下,萘接近完全轉化的溫度約為200 ℃;而在5.0 MPa時,萘接近完全轉化的溫度升至400 ℃。
從圖7b可看出,在該體系中,對應每個壓力,隨溫度的升高,四氫萘的平衡組成存在一個峰值,在壓力為0.1 MPa時,對應溫度約為250 ℃。隨系統壓力的升高,對應四氫萘平衡組成峰值的溫度逐漸向高溫偏移。在圖7b中,由于計算步長的設計問題,導致數據的連續性不佳,但圖內數據仍反映了四氫萘的平衡組成隨反應條件變化的趨勢。
從圖7c可看出,在該體系中,對應每個壓力,隨溫度的升高,反式十氫萘的平衡組成不斷降低。而隨壓力的升高,有利于生成反式十氫萘的溫度區域逐漸變寬,說明系統壓力的升高有利于反式十氫萘的生成。
從圖7d可看出,在該體系中,對應每個壓力,隨溫度的升高,順式十氫萘的平衡組成存在一個峰值,在壓力為0.1 MPa時,峰值對應溫度約為200 ℃;在壓力為5.0 MPa時,峰值對應溫度約為300 ℃。隨系統壓力的升高,峰值對應溫度逐漸向高溫偏移,且峰值逐漸增大。通過比較圖7c和圖7d可看出,在熱力學上萘加氫生成反式十氫萘比生成順式十氫萘有利,如在0.1 MPa下,反式十氫萘在含氫基的體系中的平衡組成最高可達約16.3%(φ),而順式十氫萘的平衡組成最高只有0.78%(φ)。
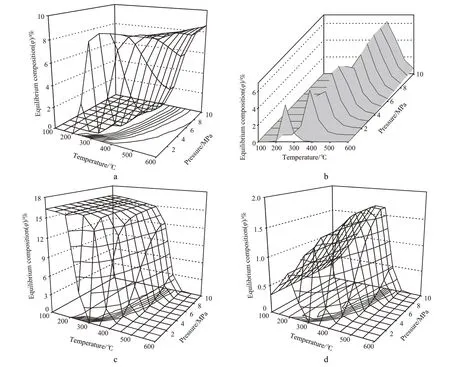
圖7 溫度和壓力對萘體系加氫產物分布的影響Fig.7 Effects of temperature and pressure on the equilibrium composition of the products in naphthalene hydrogenation.
在以氣相萘為模型反應物的芳烴加氫系統中,由于使用的催化劑不同,反應過程的活性溫度范圍可能差異很大。對于一定條件的進料,為了更好地獲得某種產物,可協調反應溫度和壓力以得到合適的操作條件。
3 計算結果與討論
本工作利用三維視圖的形式對多因素進行了描繪和考察,結果表達明了簡潔。雖然沒有進行各種芳烴及其所有加氫產物的熱力學計算,但所選幾種芳烴都是生產和實驗研究中經常使用的典型模型反應物。以上計算結果有助于研究者尋找典型芳烴加氫在熱力學上合適的操作區域,作為實驗中選擇反應條件的依據。對于實際操作過程,可以選擇它們的合適操作區域的公共部分,在熱力學上保證不同芳烴加氫過程的可行性。特別是實驗操作過程中,一般會針對反應過程選擇具體的模型反應物,而這些典型芳烴的熱力學計算會直接提供相關熱力學上所允許的反應溫度、氫烴比和系統壓力等操作條件,從而使整個研究過程得以簡化。
芳烴加氫過程的強放熱效應是由加氫過程中氫原子的加入破壞了芳烴有機物的共軛雙鍵引起的。根據化學平衡原理,隨溫度的升高,K減小,反應平衡體系中芳烴平衡組成增加,芳烴加氫產物的平衡組成減小[16]。溫度是影響芳烴加氫反應熱力學平衡的重要因素。從熱力學計算可知,芳烴加氫是可逆反應,從圖2~4和圖6可看出,溫度對芳烴加氫反應有重要影響;并且由于芳烴加氫反應是強放熱反應,平衡轉化率隨溫度的升高而降低。雖然從熱力學平衡角度低溫有利于芳烴的加氫轉化,但由于動力學的限制,實際芳烴加氫轉化率也會受到很大限制。在高溫下,芳烴加氫反應在動力學上通常會進行得非常快,但在熱力學上會受到熱力學平衡的限制,芳烴加氫的轉化率反而會降低[51]。因此,必須針對催化劑和工藝選取合適的操作溫度。在實際實驗操作和工業生產中,要充分考慮工況中的問題,如反應過程中熱點的出現會使反應床層溫度升高,從平衡角度看,必然對提高芳烴加氫的轉化率不利。因此,如果在反應過程中提高反應壓力和氫烴比,則可使保持高芳烴轉化率的可操作溫度范圍變寬,從而保證生產的順利進行。在生產過程中,催化劑會部分失活,有時需要提高反應溫度使生產進行下去,要生產低芳烴含量的油品和溶劑,則必須提高反應系統的壓力和氫烴比。
4 結論
1)利用HSC-Chemistry4.0軟件對幾種常見芳烴加氫反應進行了熱力學計算,考慮了溫度、壓力和氫烴比3個因素對芳烴加氫過程中芳烴轉化率的影響。根據化學平衡原理,隨溫度的升高,熱力學平衡常數K減小,反應平衡體系中芳烴平衡組成增大,芳烴加氫產物的平衡組成減小。
2)對于苯加氫體系,從溫度、氫烴比和壓力3個因素對芳烴加氫轉化率的三維響應面及其等高線圖可看出,等高線99.9%甚至99.0%以內的區域所對應的溫度、氫烴比和壓力為芳烴加氫高轉化率的較佳操作條件。
3)在萘加氫體系中,產物較為復雜,對應不同溫度和壓力,它們的含氫基平衡組成不同,各種產物的選擇性也不同。這些計算為選擇該過程操作參數和條件提供了參考和基礎數據。
[1] 李大東,蔣福康. 清潔燃料生產技術的新進展[J]. 中國工程科學,2003,5(3):6 - 14.
[2] 李大東. 我國環境友好汽車燃料的發展方向[J]. 中國石化,2002(4):22 - 25.
[3] 閔恩澤,李大東. 環境友好石油煉制技術的進展[J]. 石油化工動態,1997,5(3):6 - 11.
[4] Stull D R,Westrum E F,Sinke G C. Chemical Thermodynamics of Organic Compounds[M]. New York:Wiley,1969:235 - 400.
[5] Poling B E,Prausnitz J M,O’Connell J P. The Properties of Gases and Liquids[M]. New York:McGraw-Hill,2001:6.1 - 6.34.
[6] Shaw R,Golden D M,Benson S W. Thermochemistry of Some Six-Membered Cyclic and Polycyclic Compounds Debated to Coal[J]. J Phys Chem,1977,81(12):1716 - 1729.
[7] Stein S,Golden D M,Benson S W. Prediction Scheme for Thermodynamical Properties of Polycyclic Aromatic Hydrogenation[J]. J Phys Chem,1977,81(2):314 - 316.
[8] Frye C G. Equilibria in the Hydrogenation of Polycyclic Aromatics[J]. J Chem Eng Data,1962,7(4):592 - 595.
[9] Frye C G,Weitkamp A W. Equilibrium Hydrogenations of Multi-Ring Aromatics[J]. J Chem Eng Data,1969,14(3):372 - 376.
[10] Jaffe S B. Kinetics of Heat Release in Petroleum Hydrogenation[J]. Ind Eng Chem Process Des Dev,1974,13(1):34 - 39.
[11] 李宣文,黃志淵,譯. 接觸催化:工業催化劑原理、制備及其應用[M]. 北京:石油工業出版社,1984:7.
[12] Le Page J F. Applied Heterogeneous Catalysis:Design,Manufacture,Use of Solid Catalysts[M]. Paris:Technip,1987:78 - 80.
[13] Wilson M F,Fisher I P,Kriz J F. Hydrogenation and Extraction of Aromatics from Oil Sands Distillates and Effects on Cetane Improvement[J]. Energy Fuels,1987,1(6):540 - 544.
[14] Gully A J,Balard W P. Hydrogenation of Catalytic Cracking Charge Stocks[M]//McKetta J J,ed. Advances in Petroleum Chemistry and Refining. New York:Interscience Publishers,1963,7:241 - 282.
[15] Stanislaus A,Copper B H. Aromatic Hydrogenation Catalysis:A Review[J]. Catal Rev Sci Eng,1994,36(1):75 - 123.
[16] 李大東. 加氫處理工藝與工程[M]. 北京:中國石化出版社,2004:119 - 129.
[17] Tsai K Y,Wang I,Tsai T C. Zeolite Supported Platinum Catalysts for Benzene Hydrogenation and Naphthalene Isomerization[J]. Catal Today,2011,166(1):73 - 78.
[18] Boudjahem A G,Bouderbala W,Bettahar M. Benzene Hydrogenation over Ni-Cu/SiO2Catalysts Prepared by Aqueous Hydrazine Reduction[J]. Fuel Process Technol,2011,92(3):500 - 506.
[19] Abu Bakar N H H,Bettahar M M,Abu Bakar M,et al. Low Temperature Activation of Pt/Ni Supported MCM-41 Catalysts for Hydrogenation of Benzene[J]. J Mol Catal A:Chem,2010,333(1/2):11 - 19.
[20] Savva P G,Goundani K,Vakros J,et al. Benzene Hydrogenation over Ni/Al2O3Catalysts Prepared by Conventional and Sol-Gel Techniques[J]. Appl Catal,B,2008,79(3):199 - 207.
[21] Reshetnikov S I, Ivanov E A, Startsev A N. Benzene Hydrogenation in the Thiophene Presence over the Sulfide Ni-Mo/γ-Al2O3Catalyst Under Periodic Operation:Kinetics and Process Modeling[J]. Chem Eng J,2007,134(1/3):100 - 105.
[22] 孫書田. NCG-98H新型苯加氫催化劑的工業應用[J]. 化學工業與工程技術,2010,31(2):47 - 49.
[23] 宋華,唐龍,武顯春. 負載型Ni-B/γ-Al2O3非晶態合金催化劑的結構及苯加氫性能[J]. 現代化工,2010,30(1):54 - 56.
[24] 侯朝鵬. FC制備鎳金屬催化劑苯加氫反應和抗噻吩能力的研究[D]. 天津:天津大學,2003.
[25] 趙會吉,張津林,白銳,等. 新型固定床Raney Ni催化劑的苯加氫活性及耐硫性[J]. 石油化工,2007,36(7):653 -658.
[26] 傅送保,段燕凌,任軍,等. 鎳基均相絡合催化劑苯加氫反應動力學研究[J]. 石油化工,2006,35(8):745 - 748.
[27] 周志明,李卓,程振民,等. 在Pd/-Al2O3催化劑上氣相苯加氫反應動力學研究[J]. 石油化工,2003,32(5):392 - 397.
[28] Ito K,Kogasaka Y,Kurokawa H,et al. Preliminary Study on Mechanism of Naphthalene Hydrogenation to Form Decalins via Tetralin over Pt/TiO2[J]. Fuel Process Technol,2002,79(1):77 - 80.
[29] Ren Shibiao,Zhang Ping,Shui Hengfu,et al. Promotion of Ni/SBA-15 Catalyst for Hydrogenation of Naphthalene by Pretreatment with Ammonia/Water Vapour[J]. Catal Commun,2010,12(2):132 - 136.
[30] Du Mingxian,Qin Zhangfeng,Ge Hui,et al. Enhancement of Pd-Pt/Al2O3Catalyst Performance in Naphthalene Hydrogenation by Mixing Different Molecular Sieves in the Support[J].Fuel Process Technol,2010,91(11):1655 - 1661.
[31] Li Feng,Yi Xiaodong,Zheng Jinbao,et al. A Pretreatment Method of Ni/γ-Al2O3Catalyst for Naphthalene Hydrogenation[J]. Catal Commun,2009,11(4):266 - 271.
[32] Chen Honglin,Yang Hong,Omotoso O,et al. Ring,Contribution of Hydrogen Spillover to the Hydrogenation of Naphthalene over Diluted Pt/RHO Catalysts[J]. Appl Catal,A,2009,358(2):103 - 109.
[33] Sebastián D,Bordejé E G,Calvillo L,et al. Hydrogen Storage by Decalin Dehydrogenation/Naphthalene Hydrogenation Pair over Platinum Catalysts Supported on Activated Carbon[J]. Int J Hydrogen Energy,2008,33(4):1329 - 1334.
[34] Monteiro-Gezork A C A,Natividad R,Winterbottom J M.Hydrogenation of Naphthalene on NiMo-,Ni- and Ru/Al2O3Catalysts:Langmuir-Hinshelwood Kinetic Modeling[J].Catal Today,2008,130(2/4):471 - 485.
[35] Romero C M C,Thybaut J W,Marin G B. Naphthalene Hydrogenation over a NiMo/γ-Al2O3Catalyst:Experimental Study and Kinetic Modeling[J]. Catal Today,2008,130(1):231 - 242.
[36] Kirumakki S R,Shpeizer B G,Sagar G V,et al. Hydrogenation of Naphthalene over NiO/SiO2-Al2O3Catalysts:Structure-Activity Correlation[J]. J Catal,2006,242(2):319 - 331.
[37] 張小菲,邵正鋒,毛國強,等. 萘在貴金屬Pd、Pt 及Pd-Pt催化劑上的加氫活性及耐硫性能[J] 物理化學學報,2010,26(10):2691 - 2698.
[38] 張坤. 碳納米纖維負載Pd-Pt催化劑的萘加氫抗硫性能[J].吉林化工學院學報,2008,25(2):8 - 11.
[39] Mandreoli M,Vaccari A,Veggetti E,et al. Vapour Phase Hydrogenation of Naphthalene on a Novel Ni-Containing Mesoporous Aluminosilicate Catalyst[J]. Appl Catal,A,2002,231(1/2):263 - 268.
[40] 王錦惠,李保恩. 萘加氫制四氫萘絕熱反應器的研究[J]. 石油化工,1982,11(3):196 - 201,212.
[41] 王智強,李偉,張明慧,等. Ni2Mo3N/Hβ催化劑芳烴飽和加氫與開環性能[J]. 石油學報:石油加工,2005,21(2):23 - 27.
[42] 李洪寶,黃衛國,康小洪,等. 含氮化合物對NiW體系催化劑芳烴加氫性能的影響[J]. 石油煉制與化工,2006,37(10):27 - 31.
[43] 李洪寶,黃衛國,康小洪,等. 載體對Ni2W加氫催化劑活性相及芳烴飽和性能的影響[J]. 石油學報:石油加工,2006,22(6):69 - 75.
[44] Yasuda H,Kameoka T,Sato T,et al. Sulfur-Tolerant Pd-Pt/Al2O3-B2O3Catalysts for Aromatics Hydrogenation[J]. Appl Catal,A,1999,185(2):199 - 201.
[45] Yasuda H,Sato T,Yoshimura Y. Influence of the Acidity of USY Zeolite on the Tolerance of Pd-Pt Catalysts for Aromatics Hydrogenation[J]. Catal Today,1999,50(1):63 - 71.
[46] Rautanen P A,Aittamaa J R,Krause A O I. Liquid Phase Hydrogenation of Tetralin on Ni/Al2O3[J]. Chem Eng Sci,2001,56(4):1247 - 1254.
[47] Mhaouer M,Lemberton J L,Pérot G. Hydrogenation of Tetralin on a Sulfided Ruthenium on KY Zeolite Catalyst:Effect of the Sulfidation Method[J]. Catal Today,1996,29(1/4):241 - 244.
[48] Yasuda H,Higo M,Yoshitomi S,et al. Hydrogenation of Tetralin over Sulfided Nickel-Tungstate/Alumina and Nickel-Molybdate/Alumina Catalysts[J]. Catal Today,1997,39(1/2):77 - 87.
[49] Da Costa P,Lemberton J L,Potvin C,et al. Tetralin Hydrogenation Catalyzed by Mo2C/Al2O3and WC/Al2O3in the Presence of H2S[J]. Catal Today,2001,65(2/4):195 - 200.
[50] 王雷,邱建國,李奉孝. 四氫萘加氫裂化反應動力學[J].石油化工,1999,28(4):240 - 243.
[51] Girgis M J,Gates B C. Reactivities,Reaction Networks,and Kinetics in High-Pressure Catalytic Hydroprocessing[J].Ind Eng Chem Res,1991,30(12):2021 - 2058.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
