醫療器械包類產品技術審評規范探討
2013-11-12 07:21:40仲志真錢虹儲云高戴嗣衛汪澤朱穎峰孫旭穎
中國醫療器械雜志 2013年3期
仲志真,錢虹,儲云高,戴嗣衛,汪澤,朱穎峰,孫旭穎
上海市食品藥品監督管理局認證審評中心,上海市,200020
醫療器械包類產品(以下簡稱:器械包)是指兩種或兩種以上器械產品(至少有一個組件應為醫療器械產品)按一定的要求組合并實現特定的醫療目的成套器械及其容器的總稱。
近年來,器械包發展的速度很快,成為一種常用產品。本文主要就器械包產品的發展及現狀、技術審評中發現的一些涉及產品安全有效的現象和問題以及對應的思考等幾個方面進行初步地探討。
1 器械包現狀
1.1 器械包的產生原因及目前生產情況
器械包的產品形式最早是醫療機構根據某一特定的醫療目的,自行組配所需的醫療器械,由醫療機構的中心供應室進行組包、清洗、滅菌后供臨床使用。隨著醫療需求的不斷發展,醫療機構面臨的醫療壓力也與日俱增,僅憑自身的供應較難滿足臨床的需要,逐步尋求專業機構進行定制。醫療器械生產企業看到其中很好的市場前景,進入器械包的生產、銷售環節。根據醫療機構的需求,將不同的器械組合成包,或進一步滅菌后銷往醫療機構。
隨著醫療機構采購醫療器械越來越多采用公開招標的方式進行,出現了以招標文件需求進行組包的產品。在某種程度上刺激了醫療器械生產企業將產品打包銷售的熱情,加之進入該領域相對比較容易,所以有越來越多的企業加入到器械包生產企業的行列中來。這些企業中生產能力良莠不齊,對醫療器械生產質量管理和質量控制的能力差異也很大。
在我們日常的器械包的注冊審評中器械包的生產方式目前主要有以下三種:
(1) 器械包內的所有組件均由申報注冊的企業自身生產;
(2) 器械包內的主要組件由申報注冊的企業生產,其他的組件外購;
(3) 器械包內的所有組件均由申報注冊的企業外購。
第一種生產方式較少,第二種生產方式較多,也就是說申報器械包注冊的企業本身就是器械包中某些組件的生產者,自身有一定的醫療器械產品生產能力。第三種生產方式,企業本身不生產器械包中的任何組件,所有的組件均向其他企業采購而得,申報器械包注冊的企業僅僅是買來器械包中的組件,進行拆包裝、組包,然后再將滅菌過程外包給專門的機構進行滅菌處理,自身沒有任何醫療器械產品生產能力。以工作實踐的經驗來看,以第三種生產方式生產器械包的企業對質量管理的意識比較薄弱、對質量控制的能力一般也較弱。
1.2 器械包的種類和管理分類
隨著科學技術和醫療水平的不斷發展,器械包按照臨床需求的變化器械包品種多、規格多。目前,常用的器械包可分為,手術器械包(例如:口腔正畸包、醫用口腔包、骨科基礎手術器械包、無柄髖關節安裝手術器械包、普外科急救器械包等)、非手術器械包(例如:一次性使用敷料包、一次性使用氣管插管包、中心靜脈置管術換藥包、保健盒-血壓計和聽診器組合包裝 等)。根據用途的不同亦可以分為護理包和工具包。護理包是指為某些特定的臨床診療階段提供傷口護理的器械包,比如:中心靜脈置管術換藥包、一次性使用敷料包等。而工具包則是為臨床的特定診療階段提供使用的工具,之前提及的骨科基礎手術器械包、無柄髖關節安裝手術器械包等應為工具包之列。根據使用的特征器械包亦可以分為重復使用和一次性使用兩大類,在以上的舉例中,骨科基礎手術器械包、無柄髖關節安裝手術器械包、普外科急救器械包、保健盒-血壓計和聽診器組合包裝一般是重復使用器械包;而一次性使用敷料包、一次性使用氣管插管包、中心靜脈置管術換藥包可以為一次性使用器械包。
根據《醫療器械分類規則》(局令第15號)[1],凡器械包內含有III類醫療器械的,作為III類產品管理;只含有II類和I類醫療器械的,作為II類產品管理;只含有I類醫療器械的,作為I類產品管理。
1.3 國內器械包注冊現狀
依據國家局數據庫統計2009年至2011年我國境內批準上市的器械包,具體情況如下:
2009年至2011年的器械包注冊證數量2071個,從預期用途來看其中護理包占61.9%,工具包占36.3%,其它占1.8%。從使用特征來看,一次性使用產品占63.8%,可重復使用的產品占36.2%。按照管理類別,I類產品占51.7%、II類產品占42.6%、III類產品僅占5.7%,見圖1。這三年器械包的主要生產省市情況見圖2。

圖1 全國器械包注冊情況Fig.1 National medical devices kits registration situation
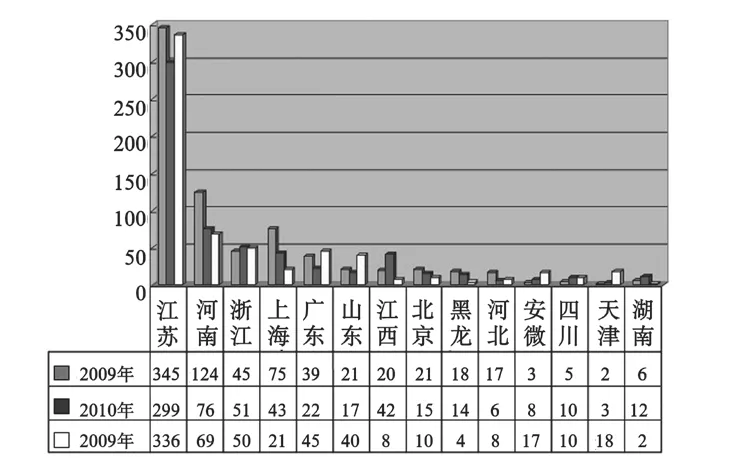
圖2 主要省市器械包生產情況Fig.2 Main provinces medical devices kits production situation
由于各省市地方審批部門在對法規把握程度和對產品標準的理解方面或多或少存在著差異,加之器械包產品的覆蓋的專業類別廣泛,所以也存在著難于統一審評尺度的現狀。
目前,上海地區有I,II類器械包生產企業約36家,I類有效注冊證77張,II類有效注冊證54張,III類有效注冊證35張(截止到2011年12月)。近三年上海所獲注冊證數量見圖3。
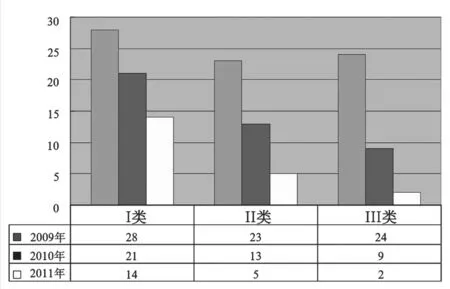
圖3 上海市器械包注冊情況Fig.3 Shanghai medical devices kits registration situation
從預期用途來看其中護理包占34.5%,工具包占61.8%,其它占3.7 %。從使用特征來看,一次性使用產品占38.9%,可重復使用的產品占61.1%。按照管理類別,I類產品占45.3%、II類產品占29.5%、III類產品僅占25.2%。
1.4 器械包內組件的情況
器械包中組件的情況呈現多樣性,基本為以下幾種:
(1) 包內組件均屬醫療器械產品,均具有有效醫療器械注冊證(以下簡稱:有證);
(2) 包內組件均屬醫療器械產品,部分組件尚未經注冊。
(3) 包內組件均屬醫療器械產品,均未經注冊;
(4) 包內組件部分屬醫療器械產品,部分為非醫療器械產品,屬醫療器械產品的組件均具有有效醫療器械注冊證;
(5) 包內組件部分屬醫療器械產品,部分為非醫療器械產品,屬醫療器械產品的部分組件具有有效醫療器械注冊證,部分組件尚未經注冊;
(6) 包內組件部分屬醫療器械產品,部分為非醫療器械產品,屬醫療器械產品組件均未經注冊。
2 現有法規文件和相關標準
目前,針對器械包的法律法規和相關文件不多,截止2012年年底,國家局發布了2個有關器械包產品的技術標準,YY/T0720-2009《一次性使用產包自然分娩用》[2]、YY0321.1-2009《一次性使用麻醉穿刺包》[3]。對于器械包還沒有較明確的技術審評指導原則。
(1)北京市于2009年發布了《一次性使用器械包類產品技術審評規范(2009版)》[4];后又于2012年再次改版發布《一次性使用器械包(盒)產品技術審評規范(2012版)》[5]。在2012年的規范中針對導尿包、檢查包、護理包、備皮包、換藥包、口腔器械盒、手術敷料包等產品進行了闡述和規范。該審評規范提出對包內組件的要求如下:
①有證組件應提供醫療器械注冊證;
② 未經注冊的醫療器械組件應對其性能提出要求;
③其他器械應明確性能要求或提供合格證明。對含有消毒劑的產品,應有提供消毒劑衛生許可證的要求,消毒劑濃度及含量的要求,消毒效果的要求。
(2) 遼寧省在2008年5月21日對本省發布了《一次性使用無菌手術包技術指導原則》[6],該指導原則適用于以非織造布及醫用脫脂紗布為主要原料加工制成的一次性使用無菌手術包(以下簡稱手術包),該產品于臨床手術中使用。
(3) 上海市在執行醫療器械相關法規要求的基礎上,對器械包的申報資料和注冊技術審評提出了如下具體的要求:
①包內組件的要求
對于包內組件已取得有效醫療器械產品注冊證,其主要技術要求應作主要參數寫入器械包產品注冊標準中。
對于包內組件包含未取得有效醫療器械產品注冊證或非醫療器產品組件,需在產品標準中明確該組件的全部技術要求和相應檢驗方法。
② 組件的用途
有注冊證產品的用途與器械包內特定用途不完全一致時,不應簡單地作為有證產品在此予以認可,應由技術審評根據具體技術要求予以審查。
③有注冊證組件的二次消毒或滅菌
企業首先需要確認涉及二次消毒、滅菌的組件。在組包前需要進行二次滅菌的驗證。包內有注冊證產品如為一次性使用的無菌產品、或有微生物控制要求的產品的,原則上不可使用半成品,應采購和使用其完整包裝產品。對于部分組件拆除包裝后如不改變產品使用性能的,允許拆除原包裝后組包,但必須做好拆包分裝記錄以便溯源。
④ 組包意見及臨床資料
I、II類器械包均應提交組包意見,組包意見必須包含以下內容:該器械包的用途;該器械包的組件,及各組件的特定用途;需給出“組包是否合理,臨床是否適用、有效”等明確的意見。
I類器械包,所提交的“組包意見”應有2家二級甲等以上醫院或者相應的專科醫院的專家簽字,并經其所在醫院或科室蓋章認可。簽字專家應留有必要的聯系方式以便咨詢和核查。
II類器械包,所提交的“組包意見”應有2家二級甲等以上醫院或者相應的專科醫院的專家簽字,并經醫院蓋章認可;若包內含有未經注冊醫療器械組件的,如何提交臨床資料,應執行《醫療器械注冊管理辦法》(局令第16號)[7]附件12中的相應要求。
3 關于注冊技術審評有關問題的探討
3.1 器械包的產品標準
在器械包的產品標準中,體現包內器械的相關信息是十分重要的。對于I、II、III類器械包來說,注冊時均需要提交產品標準。在器械包產品標準中應明確列明器械包內的每一種組件的名稱、采用的原材料、型號、規格、數量和醫療器械注冊證的情況。
(1) 對于已單獨獲注冊證的有證組件的狀態與其注冊證完整、一致的情況下,申報注冊的產品標準中應明確該組件采用的是有證產品及該組件的主要技術參數、采用的原材料、具體的型號規格等信息;
(2) 對于沒有取得有效醫療器械注冊證的醫療器械組件,企業應在器械包的產品標準中明確該組件的結構、組成,提出該組件全部的技術要求及試驗方法,若組件有國家或行業標準的,應執行相應的標準,若沒有標準,則企業應科學合理的規定該組件的全部技術要求和相應的檢驗方法;
(3) 對于包內組件非醫療器械組件,在器械包的產品標準中明確該組件的結構、組成或應執行相關的國家(行業)標準,根據其功能性來確定相關的技術要求及試驗方法。
目前在技術審評實踐中發現,企業在進行器械包注冊申報時,同一種用途的器械包會有多個不同的配置(不同組件的數量、同一組件不同的數量等情況)。器械包的不同的配置應在產品標準中予以明確。同時,在《醫療器械產品制造登記表》中應將所審批的器械包的每一種配置情況固定下來。以避免企業在器械包獲批準上市后隨意變更配置,同時監管依據也比較清晰。
3.2 器械包內有證組件的二次消毒或二次滅菌問題
器械包中有的組件在組包前已進行過一次消毒或滅菌過程,在組包后整個器械包產品送消毒或滅菌處理。對于該組件來說,其經歷了兩次消毒或滅菌的過程。這種二次消毒或滅菌對于該組件來說會產生怎樣的影響?
對于包內有證組件為一次性使用的無菌產品、或有微生物控制要求的產品的,采購來時組件已經過消毒或滅菌過程,無論組包生產時是否進行拆除包裝,企業在組包前需進行技術評估和驗證以確認這種二次消毒或滅菌對于該組件使用性能、安全性的影響。在組包生產過程中,需對有證的無菌或有微生物控制的組件進行拆包裝工序的,企業必須做好拆包分裝記錄以便溯源。
3.3 如何確認器械包內組件“有證”的問題
(1) 器械包內外購的“有證”的組件,其注冊證中明確為消毒或無菌的單包裝,但是器械包生產企業采購來的是非消毒或滅菌的大包裝,企業進行拆包裝,再組包。此種情況下的該組件是否還能夠看作為有證產品,這個問題值得我們思考。
在上述情況下,經過拆大包裝或中包裝的組件僅僅是半成品而已,與有證產品比較,缺少產品包裝過程和一個滅菌或消毒的過程,該組件不能被簡單的視作為有證組件。器械包生產企業應對該半成品組件的生物負載進行測量,并將之列入評估消毒或滅菌時需要考慮的參數;同時對于增加的生物負載應考慮該半成品組件在消毒或滅菌后可能增加的熱原等不良因素。
(2) 器械包中“有證”的組件,其在器械包中起到的作用與該組件的注冊證中所批示的臨床預期用途不一致時,該組件是否還能夠被視作為“有證”組件?
當器械包中“有證”的組件,在器械包中起到的臨床用途與該組件的注冊證中所批示的臨床預期用途不一致時,企業不應將該組件作為“有證”產品組件。技術審評時需要按照其實際的臨床用途對其進行技術評價,并應在器械包標準中規定該組件與其臨床用途相適應的全部技術指標和相應的試驗方法。
3.4 器械包證后包內組件的替換
當器械包在獲得注冊批準后,若企業對器械包中某些醫療器械組件的采購供方發生變化企業是否需要向醫療器械主管部門提交備案說明?是否會涉及《醫療器械注冊管理辦法》(局令第16號)中有關重新注冊條款(第三十四條)的規定?
以上的這種情況在實際中屢見不鮮,但是不宜簡單的認為僅是器械包組件的供方發生改變。這需要企業進行技術評估替代組件在原材料、產品特征(結構及組成等)、主要技術參數、臨床用途等方面是否能夠達到原先組件的水平。若器械包中的起主要臨床作用的有證組件發生替代的,則需要企業對其安全性指標和主要的有效性指標進行驗證,確認后方可進行替換。
3.5 器械包中直接接觸患者非醫療器械組件的審評要點
對于器械包內直接接觸患者的非醫療器械組件,如滑石粉、口杯等,它們的安全性如何考慮?
上述組件,因直接接觸患者,企業需要對該組件的毒性重點評價。企業應在產品標準中明確該組件所采用的原材料、物理和化學特征,并應根據這些組件與人體接觸的方式、接觸的部位、接觸的時間,對該組件的生物相容性作出全面的評價。
3.6 器械包中含消毒劑組件的審評要點
在器械包中含消毒劑的組件不在少數,例如:碘伏棉球、酒精棉棒、酒精棉球等。對于消毒劑我國有明確的規定,消毒劑應取得《國產消毒劑和消毒器械衛生許可批件》。在此類器械包的注冊申報資料中,器械包生產企業必須提交該消毒劑的相關資質證明。技術審評時應了解器械包中消毒劑是否取得這樣的許可。同時應注意消毒劑的用途、使用的范圍、方法和注意事項等內容應與《國產消毒劑和消毒器械衛生許可批件》批準的內容一致進行審核。
3.7 具有醫療器械注冊證的器械包中的某一組件是否可以作為“獨立有證”的“產品”
當一個器械包在獲得醫療器械注冊批準后,其中的某一組件是否能夠作為“有證”的組件被包入其他器械包中?
一個器械包獲得醫療器械注冊證時,原先包內的非有證組件的用途、使用方法等等,是依據該組件在整個器械包中的使用過程中的作用進行技術審查的,該組件在另一種組包形式下,臨床用途和使用方法等可能與原有證的器械包不完全相同,因此不能因為原整個器械包已經注冊而認為某一組件就是“有證”。當該組件作為其他器械包的組件時,應該仍然被視為無證的醫療器械組件進行技術評價。
3.8 器械包的有效期
器械包中的組件可能會有自己的有效期,比如:消毒劑等,對于如何確定器械包的有效期顯然是個比較棘手的問題。目前企業沒有過多的考慮此類問題,器械包生產企業需要對整個器械包有效期進行驗證;具體到每種組件都需要考慮有效期的驗證,最后以組件最短有效期的作為器械包的有效期。
3.9 無任何包內組件生產能力的企業從事“器械包”組包生產
在現實情況中有些器械包生產企業用于組包的組件全部外購,申報器械包注冊的企業僅對買來的組件進行拆包裝,組包,或再將滅菌過程外包給專門的機構進行滅菌處理。此種“器械包生產企業”的生產方式能否能理解為“生產”?
這些企業的生產方式與其說是“生產”,還不如說其更接近于“經營”行為。企業自身對產品的理解、對產品的質量管理的意識和質量控制的能力均很薄弱,企業對生產過程中對可能產生的風險認識也不足。這給器械包產品本身會帶來很多的不確定性。但目前在法規層面對這類生產企業沒有很清晰的要求,有待今后法律法規的完善。
3.10 器械包類產品的臨床資料問題
根據《醫療器械注冊管理辦法》(局令第16號)的要求,I類器械包不需要提交臨床資料。II類器械包需要提交臨床資料。但是法規中找不到對于器械包這一特殊產品的具體要求。
器械包內的組件在絕大部分情況下均是常見的器械,器械包本身的臨床用途是這些組件協同配合的結果,所以器械包本身的組包是否合理是臨床考慮的關鍵。因此,無論是I類器械包還是II類器械包都應在注冊申請時提交由醫療機構出具的組包意見。若器械包內的II類醫療器械組件為“無證”產品,且不在國家食品藥品監督管理局出臺的臨床豁免目錄中,則需要企業提供該組件的臨床資料或整個器械包的臨床資料。
4 結語
在醫療器械的注冊技術審評中,器械包產品與單一的醫療器械在審評的原則和思路上并沒有很大的區別,但是器械包產品在具體的技術審評尺度把握上容易出現分歧。筆者在實際的工作中遇到了一些問題希望能夠從現有的法規層面上得到解決之道,但是結果差強人意。所以這篇文章是基于將我們在實際工作中的困惑和思考呈現出來,如何在安全、有效的兩大原則基礎上使得器械包的注冊技術審評更加完善,使得器械包在上市后更加規范、有序。由于作者的能力有限,作此文章完全希望能引起從事相關工作的同行們的注意,以此拋磚引玉,使器械包注冊技術審評工作得以更好的開展。
[1]國家藥品監督管理局.醫療器械分類規則(局令第15號)[S].2000.
[2]國家食品藥品監督管理局.YY/T 0720-2009 一次性使用產包 自然分娩用[S].
[3] 國家食品藥品監督管理局.YY 0321.1-2009 一次性使用麻醉穿刺包[S].
[4] 北京市藥品監督管理局.一次性使用器械包類產品技術審評規范 2009版[S].
[5] 北京市藥品監督管理局.一次性使用器械包(盒)產品技術審評規范(2012版)[S].
[6] 遼寧省食品藥品監督管理局技術審評中心.一次性使用無菌手術包技術指導原則[S].2008.
[7] 國家食品藥品監督管理局.醫療器械注冊管理辦法(局令第16號)[S].2004.
猜你喜歡
當代水產(2022年8期)2022-09-20 06:44:30
現代裝飾(2022年4期)2022-08-31 01:39:32
現代裝飾(2022年3期)2022-07-05 05:55:06
當代水產(2022年6期)2022-06-29 01:11:44
當代水產(2022年5期)2022-06-05 07:55:06
當代水產(2022年3期)2022-04-26 14:27:04
當代水產(2022年2期)2022-04-26 14:25:10
云南畫報(2020年9期)2020-10-27 02:03:26
Coco薇(2015年1期)2015-08-13 02:23:50
玩具(2009年10期)2009-11-04 02:33:14
