乙酰乙酸乙酯合成1,1-二苯基-1-丁烯-3-酮的研究
2013-10-22 06:13:08劉希慧
唐山學院學報 2013年6期
劉希慧,卓 馨
(1.安徽理工大學 化學工程學院,安徽 淮南232001;2.自旋電子與納米材料安徽省重點實驗室,安徽 宿州234000)
乙酰乙酸乙酯是典型的β-酮酸酯,具有羰基、活性亞甲基和酯基的反應特性,可轉變為多種類型的化合物,是有機合成中的重要中間體,目前利用乙酰乙酸乙酯已合成了許多物質[1-2],乙酰乙酸乙酯乙二醇縮酮就是其中的一種物質,它具有蘋果的香氣,常用在日化香精中,一般用質子酸或者路易士酸催化乙酸乙酯與醇的方法進行合成[3-4]。α,β-不飽和酮數目眾多,它含有1,2-碳氧羰基與3,4-碳碳雙鍵形成的1,4-共軛體系,具有很好的反應活性,是有機合成中重要的原料和關鍵的中間體,已被廣泛應用于眾多領域,例如醫藥、農藥、香料等[5]。目前已報道了多種合成α,β-不飽和酮的方法[6-9],但近幾十年來,化學工作者仍一直對α,β-不飽和酮的合成研究進行改進和創新,期望得到更簡便、經濟、綠色的合成方法。在此背景下,本文介紹了一種在格氏試劑的存在下,利用乙酰乙酸乙酯合成1,1-二苯基-1-丁烯-3-酮的方法。
1 實驗部分
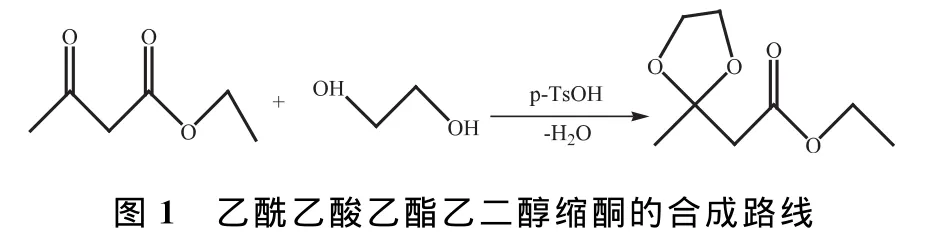
1.1 乙酰乙酸乙酯乙二醇縮酮的制備
50mL圓底燒瓶中加入150mg(0.9mmol)一水合對甲苯磺酸催化劑、3.0mL乙酰乙酸乙酯、3.0mL乙二醇和20 mL無水甲苯,裝上水分離器和回流冷凝管,分水器中注滿無水甲苯,磁力攪拌條件下油浴回流約2h,待分水器中的水層不再增加,甲苯層從渾濁變為清澈,冷至室溫時,先用10mL 5%NaOH溶液洗滌反應液,再用20mL水洗滌2次,仔細分出甲苯層,用無水硫酸鎂干燥,將干燥后的甲苯過濾到50 mL圓底燒瓶中,用少量無水甲苯洗滌漏斗中的干燥劑,常壓蒸餾除去甲苯溶劑,燒瓶中留下淺黃色液體即為粗產物乙酰乙酸乙酯乙二醇縮酮(簡稱縮酮酯),升高油浴溫度,減壓蒸餾,收集110~116℃/3332Pa餾分。乙酰乙酸乙酯乙二醇縮酮的合成路線如圖1所示。
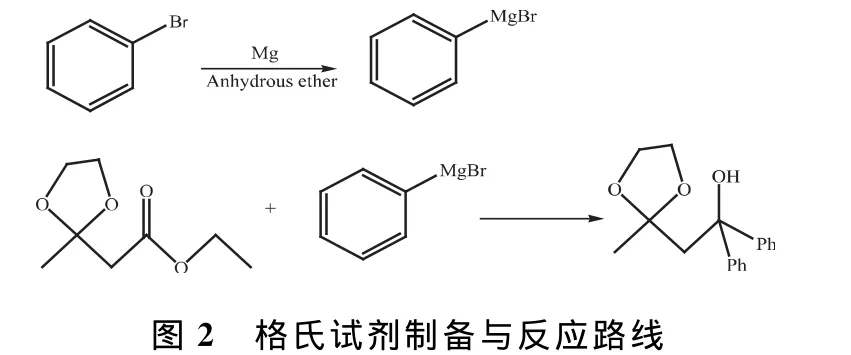
1.2 格氏試劑制備與反應
50mL三口圓底燒瓶中加入0.3g鎂屑、一小粒碘晶體和5mL無水乙醚,立即裝上滴液漏斗和帶干燥管的球形冷凝管。1.3mL溴苯溶液與5mL無水乙醚混合,通過滴液漏斗加入反應瓶中,待碘的紫色消失,溶液由黃色至乳白色時,逐滴加入溴苯溶液至乙醚沸騰,使反應液處于回流狀態,并開啟電磁攪拌使加料均勻。溴苯溶液加完后,用少量無水乙醚淋洗滴液漏斗并將其注入反應液中,攪拌下繼續油浴加熱1h,然后稱取1.0g乙酰乙酸乙酯乙二醇縮酮,將其溶于3.0mL無水乙醚中,用同一滴液漏斗將此溶液在攪拌下滴入上述溫熱的格氏試劑溴化苯基鎂反應液中,滴畢繼續回流10min,冷至室溫。在冰水浴冷卻并攪拌下通過滴液漏斗加入12mL飽和NH4Cl溶液使反應物水解,至無氣體逸出時靜置分層,轉入分液漏斗,分出醚層,水層用乙醚萃取,合并乙醚層,再用飽和NH4Cl溶液洗滌,直至水層不對石蕊試紙呈堿性為止。乙醚層用無水MgSO4干燥,過濾掉干燥劑,盛乙醚的容器和干燥劑再用2mL乙醚淋洗,合并溶液,收集在稱過重的圓底燒瓶中蒸餾除去大部分乙醚,再用溫水浴加熱進一步蒸除乙醚,瓶中殘留的黃色油狀物即粗產物1,1-二苯基-1-羥基-3-丁酮乙二醇縮酮。加石油醚1~1.5mL,用冰水浴冷卻得到淺黃色立方晶體(m.p.90~91℃)。格氏試劑制備與反應路線如圖2所示。
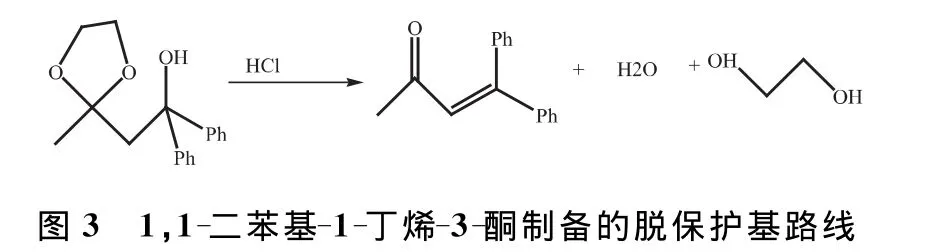
1.3 1,1-二苯基-1-丁烯-3-酮的制備
稱取上步反應產物1,1-二苯基-1-羥基-3-丁酮乙二醇縮酮,將其置于25mL圓底燒瓶中,加入1mL 4mol/L的HCl、10mL丙酮和攪拌子,裝上回流冷凝管,在攪拌下將反應混合物用油浴溫回流1h,降至室溫,再加入1mL水稀釋,用10mL乙醚分2次提取,分出乙醚提取液,依次用等體積飽和NaHCO3和水洗滌。乙醚液用無水Na2SO4干燥,將干燥后的乙醚液轉移到50mL圓底燒瓶中,干燥劑用5mL乙醚淋洗后合并到瓶中,用旋轉蒸發儀蒸出乙醚,瓶中殘留物即為1,1-二苯基-1-丁烯-3-酮粗產物。稱取重量。1,1-二苯基-1-丁烯-3-酮制備的脫保護基路線如圖3所示。
將粗產物用柱層析純化:以不同比例的石油醚與乙酸乙酯作展開劑,用硅膠薄層板法確定柱層析洗脫液組成,用50倍粗產品重的硅膠裝柱,上樣后以配好的洗脫液淋洗,收集各組分,其中主要的兩個組分分別濃縮。
2 結果與討論
在第一步的乙酰乙酸乙酯乙二醇縮酮的制備過程中,先通過常壓蒸餾除去大部分的有機溶劑甲苯,得到了粗產物,然后采用水泵進行減壓蒸餾。在減壓蒸餾的過程中先減壓至穩定,然后逐漸升溫,使壓力降一半,沸點降10~15℃。在制備過程中,采用的是甲苯和水共沸,使用分水器進行分離產物水,促使反應向正反應方向進行,當觀察到無水滴從甲苯層穿過時,即反應終止。上步反應通過減壓蒸餾,收集110~116℃時的餾分,純化得到0.7g純產物。
第一步反應的理論值:3mL×1.0 283g/mL÷130.15×174.20=4.13g;
第一步反應的產率:0.7÷4.13×100%=16.9%。
在格氏試劑的制備過程中,尤其要注意保持無水無氧的外界條件,實驗中用乙醚蒸氣作保護氣避免氧氣、水汽的影響。制備過程中,反應一旦啟動,會放出大量的熱,隨后,加熱使反應保持回流狀態。在該過程中可能會發生副反應,產生聯苯,但通過使用石油醚進行重結晶,得到了相對較純的淺黃色立方晶體:0.613g。
第二步反應的理論值:1.0g÷174.2×285=1.636g,
第二步的產率:0.613÷1.636×100%=37.5%。
第三步使用 HCl溶液將1,1-二苯基-1-羥基-3-丁酮乙二醇縮酮進行脫保護基與脫水。由于最終產物熔點低,得到的為黃色液體狀物質。對粗產物進行點板,證明有相應終產物生成。之后使用柱層析對產物進行分離。柱層析過程是利用純石油醚裝柱,用石油醚∶乙酸乙酯(30∶1)為洗脫劑進行的,在柱層析的過程中用薄層板實時檢測洗脫情況。收集目標餾分,并進行合并濃縮,最終得到黃色液體產物:0.44g。
第三步反應的理論值:0.613g÷285×222=0.478g,
第三步反應的產率:0.44÷0.478×100%=92.1%。
3 結論
本實驗在一水合對甲苯磺酸的催化下,利用乙酰乙酸乙酯與格氏試劑反應,生成了1,1-二苯基-1-丁烯-3-酮粗產物,并對其進行了分離,實驗操作簡單,過程容易控制,副產物較少且易分離。在柱層析的過程中往往要利用薄層板實時檢驗洗脫的情況,本實驗中產物為黃色物質,可以通過目測顏色來分離產物,柱層析過程簡單。
[1]丁盈紅,譚莉萍.硫酸鐵銨催化合成乙酰乙酸乙酯乙二醇縮酮[J].廣東藥學院學報,2002,18(2):81-82.
[2]袁先友,張敏,陽年發,等.微波輻射活性炭負載雜多酸催化合成乙酰乙酸乙酯縮酮[J].日用化學工業,2005,35(3):201-203.
[3]劉星明,袁先友.微波促進活性炭負載單質碘催化合成乙酰乙酸乙酯縮酮[J].湖南科技學院學報2008,29(8):47-50.
[4]北京日用化學工業學會.化工產品手冊[M].北京:化學工業出版社,1989:361-362.
[5]孫保國,何堅.香料化學與工藝[M].北京:化學工業出版社,2004.
[6]Krystyna B S,Artur B.Synthesis of phenylthio substituted 4H-pyrans and 2-pyridinones by conjugate addition-cyclization of CH-acids toα,β-unsaturated ketones[J].Monatshefte für Chemie,1999,130:545-554.
[7]Ramin G V,Hojat V.The michael addition of indoles and pyrrole toα-,β-unsaturated ketones and doubleconjugate 1,4-addition of indoles to symmetric enones promoted by pulverization-activation method and thiamichael addition catalyzed by wet cyanuric chloride[J].Mol Divers,2010,14:385-392.
[8]Chan S C,Hyo B K.Palladium-catalyzed carbonylative cyclization of b-bromo-a,b-unsaturated ketones leading to alkylidenefuranones[J].Catal Lett,2010,140:116-120.
[9]Ghorbani-Vaghei R,Hajinazari S,Engashte M.Poly(N,N′-dibromo-N-ethyl-benzene-1,3-disulfonamide),N,N,N′,N′-tetrabromobenzene-1,3-disulfonamide as new reagents for conjugate addition of indole,pyrrole withα,β-unsaturated Ketones[J].J Iran Chem Soc,2012,9:655-660.
