催化劑上環(huán)戊二烯的選擇加氫反應
2013-10-13 09:16:58梁麗萍陳霄榕
河北工業(yè)大學學報 2013年4期
關(guān)鍵詞:催化劑
梁麗萍,陳霄榕
(河北工業(yè)大學 河北省綠色化工與高效節(jié)能重點實驗室,天津 300130)
環(huán)戊烯為許多精細化工產(chǎn)品的中間體,可以用于生產(chǎn)鹵代環(huán)戊烷[1]、環(huán)戊酮[2-3]、戊二醛[4]等附加值更高的產(chǎn)品,廣泛的應用于醫(yī)藥、石油工業(yè)、食品工業(yè)等領(lǐng)域[5].但環(huán)戊烯的直接來源有限,由環(huán)戊二烯選擇加氫生產(chǎn)環(huán)戊烯最具工業(yè)應用前景.
單金屬催化劑Pd/-Al2O3加氫活性良好,但對于環(huán)戊二烯選擇加氫反應來說,選擇性稍差,容易進行深度加氫.在催化研究領(lǐng)域,添加適當助劑可調(diào)節(jié)活性組分間的相互作用,改善表面的物化性質(zhì),從而顯著改善催化劑的性能.催化劑活性組分鈀由于含有空的f軌道,對含有孤對電子的鉛有較強的吸附作用,使得鉛不易流失[6].故本課題采用鉛對催化劑Pd/-Al2O3進行修飾,制得雙金屬催化劑Pd-Pb/-A l2O3,并考察其在環(huán)戊二烯選擇加氫反應中的影響因素.
1 實驗部分
1.1 催化劑及試劑的制備
用等體積浸漬法制備雙金屬催化劑.
本課題以 -Al2O3為載體.首先將市售的 A l2O3研磨至20~40目后在550℃的條件下焙燒3 h,得到 -A l2O3.再用質(zhì)量分數(shù)為1%的鹽酸溶液分別溶解PdCl2和PbCl2,然后將載體先浸入PdCl2溶液中,陳化12 h后在120℃條件下烘干.再將Pd/-A l2O3浸入PbCl2溶液中10 h,然后在同樣條件下烘干.最后將Pd-Pb/-A l2O3在500℃下焙燒3 h.
對雙環(huán)戊二烯(DCPD)進行液相解聚:將市售的DCPD經(jīng)精餾裝置解聚,收集36~42℃之間的鎦分,得到環(huán)戊二烯(含量gt;98%).由于CPD在室溫下容易自聚成DCPD,故CPD需每次實驗前新鮮制取.
1.2 實驗流程
反應裝置如圖1所示.將催化劑裝入反應管中,在一定溫度下的氫氣流中預還原2h.待反應管冷卻到反應溫度時將環(huán)戊二烯打入反應管中,并在液空速為6 h1,氫烴比為1.0的條件下反應.產(chǎn)物經(jīng)冷凝器冷凝后進行定量、定性分析.
1.3 試樣分析
用Trace DSQ色譜-質(zhì)譜聯(lián)用進行定性分析,色譜條件如下:色譜柱為CPSIL-5(30m×0.25mm×0.12 m);分流比50∶1.質(zhì)譜條件如下:質(zhì)量數(shù)范圍為10~510 m/z;EI源溫度200℃;倍增器電壓70 eV.
用SP-1000氣相色譜儀進行定量分析.色譜條件如下:FID檢測器,50mSE-30毛細管柱(內(nèi)徑0.32 mm,膜厚0.5 m).使用N-2000型色譜工作站處理數(shù)據(jù),選用CPD的轉(zhuǎn)化率及CPE的選擇性作為催化劑活性的評價指標,采用面積歸一法計算.具體計算公式如下.
環(huán)戊二烯轉(zhuǎn)化率的計算公式為


式中:A為峰面積;f為摩爾校正因子;其中下標1代表環(huán)戊二烯,2代表環(huán)戊烯; =3,4,5,6代表副產(chǎn)物,3為環(huán)戊烷,4為雙環(huán)戊二烯,5為二氫雙環(huán)戊二烯,6為四氫雙環(huán)戊二烯.
環(huán)戊烯選擇性計算公式:

由表1可知,由于各物質(zhì)摩爾校正因子均接近1,而且副產(chǎn)物的含量較少,故摩爾校正因子均按1計算[7].

表1 各物質(zhì)摩爾校正因子表Tab.1 Mole correction factorofeach substance
1.4 催化劑表征
采用XRD對載體 -Al2O3進行物相分析,所用的儀器為荷蘭帕納科公司生產(chǎn)的X'pertPro型多晶粉末衍射儀.儀器操作條件為:Cu K 輻射( =0.154 06 nm),管電壓為40 kV,管電流為40mA,步進角度為0.02°,掃描速度為8°/m in,掃描范圍2 為10°~80°.
使用 ICP測定催化劑中活性組分鈀和修飾劑鉛的含量.所用儀器為電感耦合等離子體原子發(fā)射光譜儀(Optima ICP 7300V),工作氣體為氬氣.
使用M icromeriticsASAP 2020V3.01H型比表面與孔隙度儀測定催化劑表面參數(shù)(BET法):先將0.2g左右的樣品在真空條件下處理(200℃,3 h),然后在液氮冷阱中進行N2低溫吸附和脫附實驗.
使用M icromeritics AutoChem II2920化學吸附儀測定催化劑的還原性(TPR法):稱取0.2 g左右的樣品放入石英管中,在N2-H2混合氣氛中對實驗系統(tǒng)進行吹掃,待基線吹平后,開始程序升溫:從室溫以5℃/min的速率升至500℃.
2 實驗結(jié)果與討論
2.1 載體的物相分析
2.2 鉛鈀比對反應的影響
Pd為催化劑活性組分,添加第二金屬Pb作為修飾劑后,由于Pd和Pb的相互作用,使得催化劑表面的物化性質(zhì)、電子結(jié)構(gòu)、幾何結(jié)構(gòu)等均發(fā)生變化,催化劑性能也相應發(fā)生變化,故而鉛鈀比對反應活性有直接影響.為確定合適的鉛鈀比,分別制備鉛鈀物質(zhì)的量之比分別為0.5、0.7、1.0、1.2、1.5及未添加鉛的催化劑,并在一定條件下反應,得到CPD轉(zhuǎn)化率和CPE選擇性的關(guān)系,如圖3所示.
由圖得知,與單金屬催化劑相比,雙金屬催化劑雖然使CPD轉(zhuǎn)化率有所下降,但CPE選擇性相對提高.當鉛鈀比大于0.5時,隨著鉛鈀比的增加,CPD轉(zhuǎn)化率和CPE選擇性均呈現(xiàn)先升高后降低的趨勢.當鉛鈀比為1時,催化效果最好,CPD轉(zhuǎn)化率是90.6%,CPE選擇性是84.9%.


黃小軍[6]等人對預還原后的催化劑進行XPS表征,由表征結(jié)果可知,隨著修飾劑鉛的加入,活性組分鈀的結(jié)合能從元素態(tài)(Eb=335.2 eV)逐漸向高結(jié)合能方向移動,在鉛鈀物質(zhì)的量之比為1時,達到PdO(Eb=336.3 eV)的水平.這是由于鈀和鉛發(fā)生相互作用,一方面使得鈀難以被還原;另一方面在催化劑表面形成了鈀和鉛的化合物[10];另外,還存在一些未能被氫氣還原的氧化鉛.這些物質(zhì)是沒有加氫活性的,因為它們的的存在,覆蓋了鈀的活性中心,使其數(shù)量減少,所以改性后的催化劑使CPD轉(zhuǎn)化率下降,CPE選擇性相對提高.但當鉛鈀比繼續(xù)增大時,活性中心的數(shù)量銳減,催化劑活性便降低.
采用BET法對單金屬、雙金屬催化劑的比表面積及孔容、孔徑進行測定,如表2所示.從表中可以看出,經(jīng)改性后,催化劑的孔結(jié)構(gòu)發(fā)生了變化.結(jié)合實驗結(jié)果分析得知,較小的孔容、孔徑不易于反應的進行.這是由于環(huán)狀的CPD分子體積較大,較小的催化劑孔徑不利于反應物擴散到其內(nèi)部;而且由于擴散阻力,產(chǎn)物在孔內(nèi)的停留時間也相對較長,從而引發(fā)深度加氫等副反應,反應活性受到影響.

表2 BET分析結(jié)果比較Tab.2 Compareof the BET results
用ICP對雙金屬催化劑的Pd、Pb含量進行檢測.
將催化劑研磨成粉末后稱取一定量放入燒杯中,加入新鮮配制的王水以溶解金屬.攪拌、靜置一段時間后過濾,除去未溶解的 -Al2O3.最后將濾液轉(zhuǎn)移到容量瓶中進行定容,再取1 mg/L二價鈀和鉛的標準溶液作為參照.經(jīng)檢測后發(fā)現(xiàn)樣品中Pd的實際含量為0.962%,鉛鈀比為0.994.考慮到配制樣品溶液過程中的損失,可以認為催化劑中鈀、鉛實際含量與理論含量相同,活性組分沒有損失.
2.3 還原溫度對反應的影響
在催化劑Pd-Pb/-A l2O3中,Pd是選擇加氫的活性中心,而經(jīng)焙燒后Pd是以氧化物PdO形式存在的,催化劑需要經(jīng)過 H2預還原處理數(shù)小時.我們保持其它反應條件固定不變,通過改變催化劑的還原溫度,對催化劑的活性進行了考察,結(jié)果如圖4、5所示.
從圖中可以看出,當催化劑的還原溫度為180℃時,CPD轉(zhuǎn)化率和CPE選擇性均高于另外兩組.當還原溫度為150℃、200℃時,CPD轉(zhuǎn)化率變化較大,表明催化劑的穩(wěn)定性較差.

圖4 還原溫度對CPD轉(zhuǎn)化率的影響Fig.4 Effectof reduction temperatureon the conversion of CPD

圖5 還原溫度對CPE選擇性的影響Fig.5 Effectof reduction temperatureon the selectivity of CPE
圖6 是催化劑的TPR曲線.由圖可以看出,催化劑在138℃時還原峰最強,發(fā)生還原反應的溫區(qū)主要是在80~350℃.從實驗結(jié)果和TPR圖譜可知,預還原溫度較低時,催化劑沒有被充分還原,無法形成大量的活性中心,而且由于溫度較低使得形成的被還原的金屬顆粒較小,故其穩(wěn)定性和催化活性均較差.若還原溫度過高(200℃),催化劑可能由于燒結(jié)而聚集,使得活性組分分布改變,活性中心數(shù)目減少,導致催化劑加氫活性下降.所以只有當還原溫度為180℃時,催化劑具有較穩(wěn)定的結(jié)構(gòu),活性中心分布較為均勻而且數(shù)目較多,具有較高的加氫活性.綜合考慮,180℃為最佳的還原溫度.
2.4 反應溫度對催化劑的影響
反應溫度過低,催化劑無活性或活性太低;溫度過高則導致終產(chǎn)物中環(huán)戊烷等副產(chǎn)物增多.我們對不同反應溫度進行考察,結(jié)果如圖7所示.
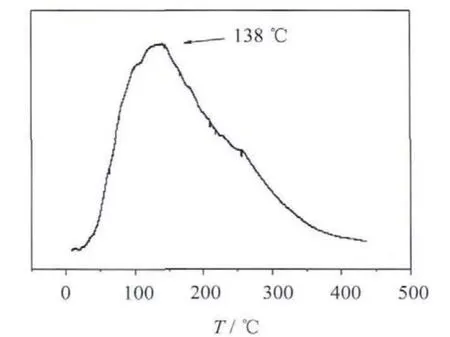
圖6 催化劑TPR曲線Fig.6 TPR profilesof catalysts

圖7 反應溫度對催化活性的影響Fig.7 Effectof tempertureon theactivity of catalyst
由圖7可知,隨著反應溫度的升高,催化劑活性也逐漸增加.當溫度從110℃升高到120℃時,CPD轉(zhuǎn)化率保持在90%左右,但CPE選擇性卻從70.7%提高到86.9%.但溫度繼續(xù)升高,CPE選擇性迅速下降.故合適的反應溫度為120℃.
這是由于溫度較低,催化劑活性組分不能使足夠的CPD分子和H2分子活化,使得加氫反應受到影響,反應速度較慢,導致催化劑活性較低.當溫度升高時,CPD轉(zhuǎn)化率和CPE選擇性均有了明顯提升.但若繼續(xù)升高到130℃,反應不僅容易出現(xiàn)飛溫現(xiàn)象,而且在高溫情況下CPD分子和H2分子完全活化,容易進行深度加氫反應,大量生成副產(chǎn)物,使得CPE選擇性下降.故合適的反應溫度為120℃.
3 結(jié)論
[1]翁羽飛,奚軍,丁仙華.鹵代環(huán)戊烷合成工藝研究進展 [J].上海化工,2004,29(10):27-29.
[2]隋超,李新勇,曲振平.環(huán)戊酮合成方法研究進展 [J].化工進展,2008,27(6):809-813.
[3]Grate John H ,Hamm David R,SaxtonRobertJ,etal.Catalytic system forolefinoxidation to carbonylproducts[P],WO91/13851:1991-09-19.
[4]朱志慶,呂自紅,范兆馨,等.環(huán)戊烯氧化合成戊二醛的催化劑的制備方法 [P],CN1911515A:2007-2-14.
[5]徐克勛.精細有機化工原料及中間體手冊 [M].北京:化學工業(yè)出版社,1998:2-15.
[6]黃小軍,李和興,戴維林,等.鉛修飾Pd/A l2O3催化劑上環(huán)戊二烯的選擇加氫反應 [J].催化學報,1997,18(5):418-520.
[7]顧蕙祥,閻寶石.氣相色譜使用手冊 [M].第2版.北京:化學工業(yè)出版社,1989,524.
[8]唐國旗,張春富,孫長山,等.活性氧化鋁載體的研究進展 [J].化工進展,2011,30(8):1756-1765.
[9]李波,邵玲玲.氧化鋁、氫氧化鋁的XRD鑒定 [J].無機鹽工業(yè),2008,40(2):54-58.
[10]Goetz J,Volpe M A,Sica A M,etal.Low-loaded Pd-Pb/-A12O3catalysts:indication of alloy formation from FYIR and X ray photoelectron spectroscopy[J].Catal,1997,167(2):314-323.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
