黃粉蟲纖溶酶的三維結構模擬與序列分析
2013-08-14 09:08:40韓雅莉
化學與生物工程 2013年9期
關鍵詞:結構
余 磊,韓雅莉
(1.武漢軟件工程職業學院,湖北 武漢430205;2.廣東工業大學,廣東 廣州510006)
黃粉蟲(Tenebrio molitor)俗稱黃粉甲、面包蟲,原產于南美洲,屬于倉庫和貯藏害蟲。因其營養成分高,營養含量居各類活體動物蛋白飼料之首,被譽為“蛋白質飼料寶庫”。作者所在課題組從黃粉蟲體內分離純化得到黃粉蟲纖溶酶[1,2],對其基因DNA序列進行了分析,克隆得到其中一種黃粉蟲纖溶酶成熟肽cDNA序列(GenBank登錄號:JN662461),并將其表達[3]。在此,利用生物信息學方法,對黃粉蟲纖溶酶序列進行多方位分析、確定活性中心的位置,建立了可靠的黃粉蟲纖溶酶三維結構模型,并揭示了黃粉蟲纖溶酶的纖溶機理。
1 方法
要想了解蛋白質的立體結構,需要將蛋白質溶液結晶,用X-射線衍射分析蛋白質的單晶才能完成。但是蛋白質結晶條件苛刻,而且不是所有蛋白質都能形成單晶,因此實施起來受到限制。利用現代計算機技術,人們可以對已知氨基酸序列的蛋白質三維結構進行預測,同時還能對蛋白質序列中的每一個氨基酸逐個分析。因此,比較蛋白質模建,又稱同源模建,逐漸成為目前應用最廣的蛋白質三維結構預測方法[4-6]。
將黃粉蟲纖溶酶的序列提交到NCBI的自動比較蛋白質模建服務器Blast上,通過程序,自動確定黃粉蟲纖溶酶序列活性中心與底物結合部位;將黃粉蟲纖溶酶蛋白質序列導入Biosun軟件,軟件自動選取蛋白質Tryp_SPc[cd00190]為模板,模擬得到黃粉蟲纖溶酶的三維結構,并利用Blast比較模板蛋白與黃粉蟲纖溶酶的同源性;利用Goldkey軟件,對黃粉蟲纖溶酶全序列等電點、親水性、柔性進行分析,基于無模型比對時活性中心氨基酸殘基的性質,進一步闡明黃粉蟲纖溶酶的纖溶機理。
2 結果與討論
2.1 黃粉蟲纖溶酶序列分析(圖1)

圖1 黃粉蟲纖溶酶序列分析Fig.1 The sequence analysis of fibrinolysin fromTenebrio molitor
由圖1可知,黃粉蟲纖溶酶序列的活性中心(Active sites)為 H72、D117、S212,底物結合部位(Substrate binding sites)為:D207、S228、G230。
2.2 三維結構模擬與評估
黃粉蟲纖溶酶的模擬三維結構見圖2,模板蛋白和黃粉蟲纖溶酶的同源性比較見表1。

圖2 黃粉蟲纖溶酶模擬三維結構Fig.2 The analogous 3D-structure of fibrinolysin from Tenebrio moliter
圖2中,a、b、c 3個球狀模型,分別對應黃粉蟲纖溶酶的活性中心S212、H72、D117三個氨基酸殘基側鏈:羥基、咪唑基、羧基,與胰蛋白酶催化三聯體結構一致[7,8]。d、e、f三處肽鍵鏈狀模型,分別對應黃粉蟲纖溶酶的底物結合部位D207、S228、G230。由三維結構可以看出底物結合部位和活性中心均沒有α-螺旋和β-折疊(α-螺旋為柱狀區,β-折疊為板狀區),該區域容易發生形變,在催化過程中與底物結合時,產生誘導契合,符合酶催化理論;酶活中心處于蛋白質中心凹穴處,與通常對纖溶酶活性中心的認識一致[9,10]。本課題組前期研究中曾發現黃粉蟲纖溶酶受絲氨酸蛋白酶抑制劑PMSF的抑制,推測其為絲氨酸蛋白酶[2,3],此推測與生物信息學方法找到的活性中心S212相吻合。
黃粉蟲纖溶酶的序列為:MKSILFVVFLVASASAVPPFLRKNSLLPDGRIVGGSSISISSVPWQISLQYYGSHICGGSIISANYIVTAAHCTDGLTAGSLTVRAGTSTRGSGGQVVNVARINQNPSYNDRLIDYDISVLQLSSSLSLGSSVAAVGLPSSSTSWSAGTSVLVTGWGTTTEGSSSLPSALQGVNVQIVSQSTCSSAYGSGSITDRMLCAGVTGGGKDACQGDSGGPLVVGNVLAGIVSWGYGCARNGYPGVYSNVPA-LRSYIQQTAGI。
模板蛋白序列為:IVGGSEAKIGSFPWQVSLQYTGGRHFCGGSLISPRWVLTAAHCVYSSAPSNYTVRLGSHDLSSNEGGGQVIKVKKVIVHPNYNPSTYDNDIALLKLKRPVTLSDNVRPICLPSSGYNLPAGTTCTVSGWGRTSEGGPLPDVLQEVNVPIVSNAECKRAYSYGGTITDNMLCAGGLEGGKDACQGDSGGPLVCNDNGRGVLVGIVSWGSGCARPNYPGVYTRVSSYLDWIQKT。

表1 模板蛋白和目的蛋白同源性比較Tab.1 The homology comparison between the template protein and the target protein
由表1可知,Blast評分為186,說明模板蛋白和目的蛋白的同源性比較高;序列覆蓋率達到了94%;隨機匹配的可能性非常小,僅為2E-62,說明兩個蛋白出現相同的序列并非偶然;最大序列的相似度達到51%。表明,以此模板蛋白進行同源模建,所得結構是比較可信的。
2.3 Goldkey軟件的序列分析
在黃粉蟲纖溶酶三維結構分析中,采用的是同源模建的方法。預測過程中,其它蛋白質與目標預測物的差異,會導致一定程度的偏差。采用Goldkey軟件對序列的每個殘基逐個計算其特性,雖然無法得到三維結構,但是其數據可靠性更好,更能反映目標蛋白質的特性。用Goldkey軟件分別對黃粉蟲纖溶酶氨基酸序列進行等電點模擬、親水性模擬、柔性模擬,結果見圖3。
由圖3a可知,黃粉蟲纖溶酶的等電點為9.16,在人體內正常生理環境下,該酶帶正電。而纖維蛋白通常帶負電荷,所以黃粉蟲纖溶酶很容易靠近纖維蛋白,并與之結合。因此,黃粉蟲纖溶酶對血栓有一定的靶向作用。
對于水溶性球形蛋白,親水值較低的氨基酸,趨向于蛋白質的內部,親水值較高的氨基酸,趨向于蛋白質的外部。由圖3b可知,H72、S212、D207親水值較低,而且H72和D207是局部親水值最低點,應該在球蛋白內部;S228、G230親水值較高,應該在球蛋白外部,與三級結構相符。
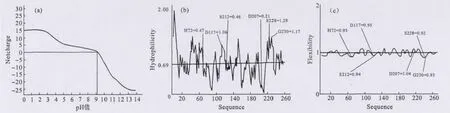
圖3 等電點模擬(a)、親水性模擬(b)和柔性模擬(c)Fig.3 The isoelectric point analogue(a),hydrophilicity analogue(b)and flexibility analogue(c)
由圖3c可知,活性部位H72、D117、S212的柔性值比較小,幾乎是整條鏈上最小的區域,說明組成酶活性中心的3個氨基酸殘基的空間位置相對固定,因而活性中心的相對結構很穩定,在自然狀態下不會隨意發生變形,底物結合部位中S228、G230的柔性值比較小,說明這兩個殘基的空間相對位置比較固定,D207的柔性值比較大,這符合酶促反應動力學誘導契合理論。在結合底物時,酶的結合部位和底物的結構都發生了一定的形變(柔性才能形變),兩者在空間結構上剛好互補,從而能很好地契合。全鏈柔性分析表明,黃粉蟲纖溶酶的活性中心和底物結合部位符合酶催化機理,從另一側面證明了活性中心預測的準確性。
2.4 纖維蛋白水解部位結構分析
體內纖維蛋白的溶解是纖溶酶作用于精氨酸-賴氨酸肽鍵,使之斷裂的過程[11,12]。
3 結論
利用生物信息學方法,模擬黃粉蟲纖溶酶的三維結構,并對其全序列進行了分析。確定黃粉蟲纖溶酶的活性中心為 H72、D117、S212,底物結合部位為D207、S228、G230。黃粉蟲纖溶酶屬于水溶性球蛋白,其纖溶活性中心位于球蛋白表面凹穴處。推測其催化纖維蛋白水解的機理是作用于精氨酸-賴氨酸肽鏈,使不溶性纖維蛋白水解成可溶性蛋白。
[1]Yu L,Han Y L,Tan Z J.Purification of a fibrinolytic protein from the yellow mealworm beetles,Tenebrio molitor[J].Advanced Materials Research,2011,396(11):103-105.
[2]陳雅雄,譚竹均,韓雅莉,等.黃粉蟲蛋白提取物纖溶活性及性質研究[J].時珍國醫國藥,2011,22(1):169-171.
[3]葉韻,韓雅莉,黃明星.黃粉蟲胰蛋白酶樣酶基因TMTLSP全長cDNA的克隆和序列分析[J].中國生物化學與分子生物學報,2012,28(2):169-176.
[4]Jaroszewski L,Rychlewski L,Li Z.A server for profile sequence alignments[J].Nucleic Acids Research,2005,33(1):284-288.
[5]Zhou H,Zhou Y.Fold recognition by combining sequence profiles derived from evolution and from depth-dependent structural alignment of fragments[J].Proteins,2005,58(2):321-328.
[6]Venclovas C,Margelevicius M.Comparative modeling in CASP6 using consensus approach to template selection,sequence-structure alignment and structure assessment[J].Proteins,2005,61(7):99-105.
[7]Rajesh S R,Chang J Y.Structural stability of human alpha-thrombin studied by disulfide reduction and scrambling[J].Biochimica et Biophysica Acta,2003,1651(1-2):85-92.
[8]Cear E D,Dang Q D,Ayala Y M.Molecular mechanisms of thrombin function[J].Cell Mol Life Sci,1997,53(9):701-730.
[9]Bode W.Structure and interaction modes of thrombin[J].Blood Cells,Molecules,and Diseases,2006,36(2):122-130.
[10]Blow D M,Birktoft J J,Hartley B S.Role of a buried acid group in the mechanism of action of chymotrypsin[J].Nature,1969,221(5178):337-340.
[11]Stubbs M T,Bode W.A player of many parts:The spotlight falls on thrombin′s structure[J].Thromb Res,1993,69(1):1-58.
[12]Swain C G,Brown J F.Concerted displacement reactions.Ⅶ.The mechanism of acid-base catalysis in non-aqueous solvents[J].J Am Chem Sci,1952,74(10):2534-2538.
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
