硼化合物催化的直接酰胺化反應研究進展
2013-08-14 09:08:30沙文彬黃文華
化學與生物工程 2013年6期
關鍵詞:催化劑
沙文彬,黃文華
(天津大學理學院化學系,天津300072)
酰胺鍵廣泛存在于天然和人工合成的化合物中。20種α-氨基酸之間利用酰胺鍵組裝形成了生命的基石——蛋白質與多肽。在很多天然或人工合成的藥物分子中,酰胺鍵是重要的連接片段。2011年,對三大國際制藥公司研發化合物的抽樣統計表明,其中54%存在酰胺鍵[1]。在一些廣泛使用的合成材料(如尼龍)中,酰胺鍵也扮演著不可替代的角色。通過酰胺的還原來制備胺類化合物也是有機合成中常見的一種轉化[2]。因此酰胺鍵的形成無論在生命活動,還是在實驗室制備和工業生產中都是十分常見的反應與過程。
目前,普遍采用的酰胺合成方法是:羧酸首先在活化試劑的作用下形成活潑中間體,如活潑酯、酰鹵和酸酐等;然后該中間體再對胺進行酰化。數以百計的此類活化試劑已經被開發出來,如碳二亞胺類、有機膦類、活潑酯類等[3,4]。雖然這些活化試劑在酰胺的制備中得到了廣泛應用,但它們的使用存在著如下的缺點:(1)這些活化試劑以及反應中添加的堿等,都要使用化學計量的,有時甚至是大為過量的;而酰化反應完成后,它們都轉變成了無用的副產物,這與當今提倡的“原子經濟性”原則相違背。(2)這些活化試劑以及它們產生的副產物往往是高毒的,給酰胺的制備和應用帶來很多困難。(3)很多活化試劑都是昂貴的。(4)這些活化試劑產生的副產物與酰胺的分離,有時是困難而繁瑣的。上述這些缺點均與“綠色化學”的理念不相符[5],尤其是在大規模制備酰胺產物時,這些缺點就更加突出。因而就不難理解2007年“避免采用低原子經濟性的試劑合成酰胺”被評選為制藥工業中的一項關鍵挑戰[6]。此外,對一些反應活性很差(如位阻很大)的底物,往往要采用劇烈的反應試劑(如將羧酸轉化為酰氯)或者需要改變合成策略,從羧酸和胺以外的底物出發來形成酰胺[7]。因此,尋找并開發高效、綠色和經濟的酰胺鍵構建方法是合成化學中亟待解決的問題[8]。硼化合物催化的直接酰胺化反應正是其中研究較多、前景最為誘人的一種方法。
作者在此對硼化合物催化的直接酰胺化反應的研究進展進行了綜述,并按照硼化合物催化劑的類型進行了分類。
1 直接酰胺化反應
不添加活化試劑,而利用羧酸和胺的直接縮合來形成酰胺(即直接酰胺化反應),可以避免使用活化試劑帶來的諸多缺陷,是合成酰胺的理想途徑。羧酸與胺是普遍而易得的底物,直接酰胺化反應的副產物只有水,而無其它廢棄物。但是羧酸與胺混合時會形成羧酸銨鹽,阻礙了酰胺的形成[9](圖1)。雖然對于直接酰胺化反應,很早就有一些報道[10,11],但均需要長時間的高溫(>160℃)加熱,且反應只適用于某些特定的羧酸或胺,使其未能推廣成為普遍采用的酰胺合成方法。
近年來出現的一系列催化劑可以使直接酰胺化反應的條件變得較為溫和,并能拓展到一般酰胺鍵的形成上。這些催化劑包括過渡金屬化合物[12]、非金屬氧化物[13]和硼化合物等,其中硼化合物是最有潛力的一類催化劑。硼化合物作為化學計量的試劑,參與促進酰胺鍵的形成很早就有報道[14,15]。而硼化合物催化的直接酰胺化反應,雖然在早期專利中有所報道,但其方法僅局限于合成特殊類型的酰胺化合物,并未引起關注[16]。自1996年Ishihara等報道取代的苯硼酸可以催化一般的羧酸和胺的直接縮合反應后,硼化合物催化的直接酰胺化反應領域才開始受到研究者的普遍關注。
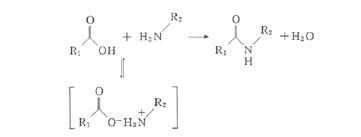
圖1 羧酸與胺直接縮合形成酰胺Fig.1 Amide from direct condensation between carboxylic acid and amine
2 硼化合物催化的直接酰胺化反應
催化劑Ⅰ可以從1-溴-3,4,5-三氟苯出發,先制成Grignard試劑,再與硼酸三甲酯反應,進而水解得到。
催化劑Ⅰ可以催化一系列羧酸與胺的直接酰胺化反應,對于反應性較差的底物(如苯甲酸、苯胺)以及位阻很大的1-金剛烷甲酸,以高沸點(165℃)的均三甲苯代替甲苯(沸點111℃)作為共沸溶劑可以得到更滿意的結果。催化劑Ⅰ的反應機理如圖3所示[18]。
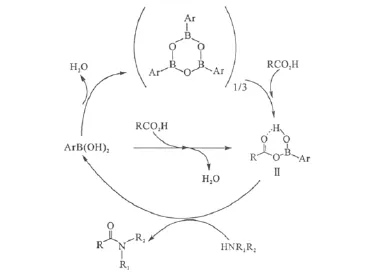
圖3 催化劑Ⅰ的可能反應機理Fig.3 Proposed catalytic principle for catalyst I
2.1 芳基硼酸作為催化劑
芳基硼酸(Arylboronic acid)具有特異的反應活性、穩定性、易得性和低毒性,是Suzuki偶聯、Chan-Lam偶聯反應的重要底物,廣泛應用于催化反應、藥物、小分子識別等領域[17]。
2.1.1 含氟取代基的芳基硼酸催化劑
Ishihara等[18,19]發現,在甲苯共沸脫水條件下,間位與對位帶有吸電子基的苯硼酸可以很好地催化直接酰胺化反應,其中3,4,5-三氟苯硼酸(Ⅰ)的催化活性最高(圖2)。
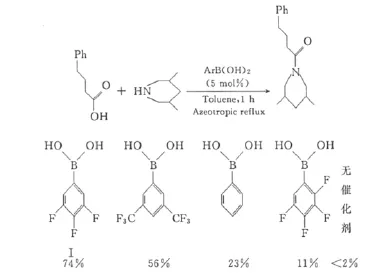
圖2 苯環上取代基對催化活性的影響Fig.2 Effect of substituents on catalytic activity
一般情況下,芳基硼酸(其中含有一定量的芳基硼酸酐)與羧酸反應可以生成單酰氧基硼酸(Ⅱ)(可視作羧酸與芳基硼酸的混合酸酐)。化合物Ⅱ中的分子內氫鍵和缺電子的硼可以活化羧基碳,有利于胺的親核進攻。
催化劑Ⅰ也可用于尼龍66和其它聚酰胺的合成。例如,以摩爾分數為10%的化合物Ⅰ作為催化劑,己二酸與己二胺在間甲酚與鄰二甲苯的混合溶劑(體積比1∶4)中回流脫水20h,可以得到數均分子量(Mn)和質均分子量(MW)分別為4690和22 400的尼龍66[20]。
2.1.2 可以重復利用的芳基硼酸催化劑
在催化劑Ⅰ的基礎上,研究者又開發了一些可以重復利用的直接酰胺化催化劑,如圖4所示。
含有全氟代烴基的催化劑Ⅲ在氟二相體系中可以很好地催化直接酰胺化反應,反應結束后,通過氟相和普通有機相的分離,可以使氟相中的催化劑Ⅲ得到完全回收和再利用[21]。
正離子芳基硼酸(Ⅳ)可以在苯甲醚、乙腈和N-甲基吡咯烷酮(NMP)等極性溶劑中使用,解決了催化劑Ⅰ無法適用于極性溶劑的缺陷。利用離子液體1-乙基-3-甲基咪唑三氟甲磺酸鹽([emim][OTf])與甲苯的二相體系可以實現催化劑Ⅳ的重復利用[22]。
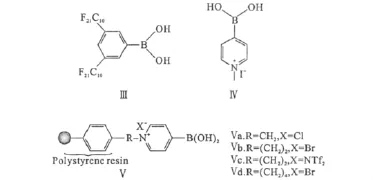
圖4 可重復利用的芳基硼酸催化劑Fig.4 Reusable arylboronic acid catalysts
與聚苯乙烯結合的催化劑Ⅴ在甲苯中反應結束后,經簡單過濾、洗滌就可以重復利用,其中Ⅴc重復利用10次后,其催化活性無明顯下降[22]。
2.1.3 氨基硼酸類催化劑
Georgiou等[23]研究了氨基硼酸類化合物對直接酰胺化反應的催化活性。以沸點較低(85℃)的氟苯作為共沸溶劑,在無催化劑的條件下,苯甲酸與芐胺或4-苯基丁胺在氟苯中除水回流并不能生成相應的酰胺;而以自行合成的氨基硼酸Ⅵ(圖5)可以有效地催化酰胺的生成,氮原子上位阻更大的Ⅵa的活性較Ⅵb要高,且二者活性都高于催化劑Ⅰ[24]。
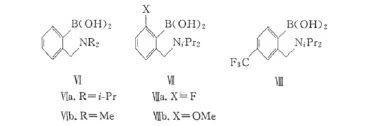
圖5 氨基硼酸類催化劑Fig.5 Aminoboronic acid catalysts
Arnold等[25]考察了苯環上不同取代基對催化活性的影響,結果發現,催化活性的順序是Ⅷ>Ⅵa>ⅦaⅦb,即當硼原子的對位引入吸電子基時催化活性會增強;而當Ⅵa中硼的另一個鄰位也被取代時,催化活性會下降,尤其是鄰位引入供電子基時催化活性會大大減弱。
2008年,Arnold等[26]報道了第一例不對稱的直接酰胺化反應。外消旋的α-甲基芐胺與非手性的羧酸在基于二茂鐵的手性氨基硼酸Ⅸ的催化下,選擇性地得到一種對映體過量的手性酰胺(圖6)。雖然,對映體的選擇性還不高,但這是第一次通過動力學拆分外消旋胺來實現不對稱酰胺化,意義重大。
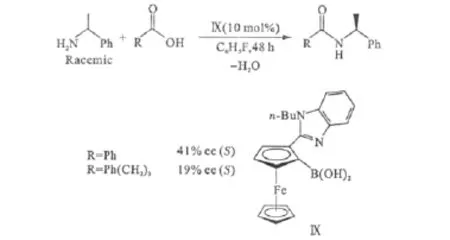
圖6 催化劑Ⅸ催化的不對稱直接酰胺化反應Fig.6 Asymmetric direct amidation catalyzed by catalystⅨ
2.1.4 鄰碘代芳基硼酸類催化劑
Al-Zoubi[27]等通過對45種芳基硼酸的篩選,發現鄰鹵代苯硼酸在以二氯甲烷為溶劑、4分子篩為吸水劑的條件下,室溫下就可以很好地催化羧酸與胺的直接縮合。其中尤以鄰碘苯硼酸(Ⅹ)的催化活性最好,且這些鄰鹵代苯硼酸的活性順序是I>Br>Cl>F(圖7)。而當硼的另一個鄰位也被鹵素取代時,催化活性會明顯下降,這一結果與Whiting等對氨基硼酸催化劑Ⅵa苯環上取代基效應的研究結果類似。
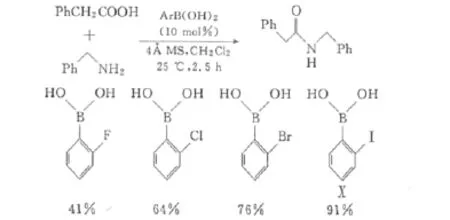
圖7 鄰鹵代苯硼酸催化活性的比較Fig.7 Comparison of catalytic activity for ortho-halophenylboronic acids
最近,Gernigon等[28]在催化劑Ⅹ的基礎上,對其結構進行了優化,以提高催化活性。首先,參照催化劑Ⅹ,在鄰鹵代苯硼酸的苯環上引入吸電子基,以提升催化活性,制備了含氟的催化劑Ⅺ,但催化劑Ⅺ的活性反而低于催化劑Ⅹ;隨后Gernigon等又合成了帶有供電子基的催化劑Ⅻ,但其催化活性與催化劑Ⅹ相比也未能提高(圖8)。
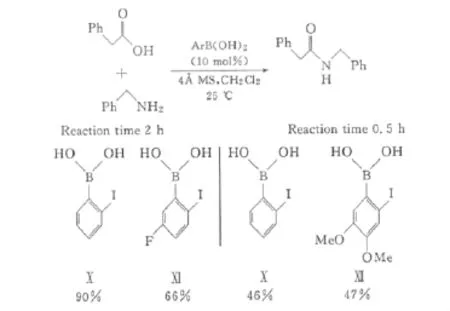
圖8 吸電子與供電子取代基對催化劑Ⅹ催化活性的影響Fig.8 Effect of electron-withdraw/-given substituent on catalytic activity of catalystⅩ
通過比較催化劑Ⅹ、Ⅺ和Ⅻ,發現在碘原子上保持較高的電子密度、同時不讓硼原子上電子密度過高,可能會提高催化劑的活性。由此合成了碘的對位帶有供電子取代基的催化劑ⅩⅢ(2-碘-5-甲氧基苯硼酸)和ⅩⅣ。研究發現它們與鄰碘苯硼酸催化劑Ⅹ相比,催化活性有了顯著提升,其中催化劑ⅩⅢ的活性最高。但若在催化劑ⅩⅢ上再引入吸電子基后,催化活性反而下降(圖9)。
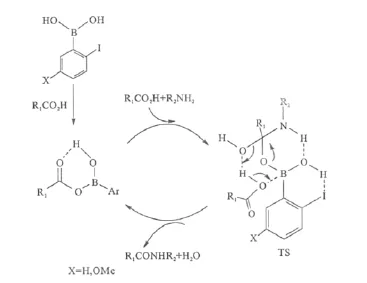
圖10 催化劑Ⅹ與ⅩⅢ的可能催化機理Fig.10 Proposed catalytic principle for catalystⅩand catalystⅩⅢ
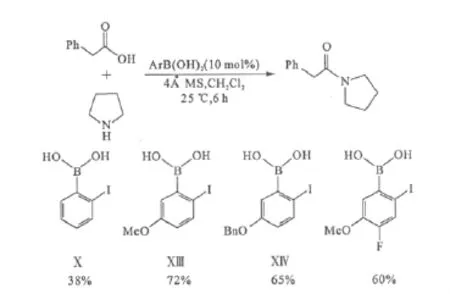
圖9 碘的對位有供電子基的苯硼酸與催化劑Ⅹ的催化活性比較Fig.9 Activity comparison between arylboronic acids with electron-given groups parato iodine and catalystⅩ
計算化學研究表明,鄰鹵代苯硼酸對直接酰胺化反應顯著的催化活性是由于其中的鹵原子作為氫鍵供體,穩定了反應過渡態;且碘參與形成氫鍵的能力要強于其它鹵素原子[29]。催化劑Ⅹ和ⅩⅢ相比,ⅩⅢ中碘原子上電子密度更高,使其形成的I-H更強,從而其參與的過渡態的能量較催化劑Ⅹ更低,因而其催化活性更高(圖10)。
2.2 硼酸作為催化劑
硼酸(Boric acid)是一種廉價易得、性質穩定、環境友好的物質,近年來作為有機反應的催化劑引起了研究者的普遍興趣[30,31]。Tang[32]報道了硼酸催化直接酰胺化反應。在摩爾分數為1%的硼酸催化下,4-苯基丁酸和芐胺在甲苯中回流分水16h,經簡單后處理可得到收率91%的N-芐基-4-苯基丁酰胺。反應機理可能是:硼酸與羧酸先形成如化合物Ⅱ的混合酸酐,再與胺反應得到酰胺,并重新生成硼酸。
硼酸作為直接酰胺化反應的催化劑,用于制備一些結構較為復雜的藥物活性成分(API)時,用量要提高到10%(摩爾分數)(圖11)[33]。
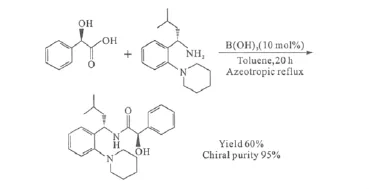
圖11 以硼酸為催化劑合成藥物活性成分(API)Fig.11 Preparation of API catalyzed by boric acid
在此過程中醇羥基并不受影響,無需保護,且底物中的手性中心也不受影響。因此,硼酸催化的直接酰胺化反應為酰胺的制備,尤其是工業上大規模制備酰胺化合物,提供了一條成本低廉、過程簡便的途徑。
此外,羧酸與鄰苯二胺在硼酸的催化下先形成酰化中間體ⅩⅤ,再縮合環化,生成2-取代的苯并咪唑類化合物(圖12)。
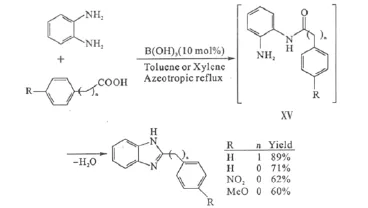
圖12 硼酸催化的2-取代苯并咪唑化合物的合成Fig.12 Synthesis of 2-substituted benzimidazoles catalyzed by boric acid
傳統上此類化合物的合成都需要在強酸性或高溫(約200℃)條件下進行,因此,硼酸催化的縮合環化為此類化合物的制備提供了一條溫和的方法,尤其適用于對酸性條件敏感的底物[34]。
2.3 其它硼化合物作為催化劑
Maki等[35]合成了基于四氯代鄰苯二酚的催化劑ⅩⅥa和ⅩⅥb。ⅩⅥa對一些大位阻的羧酸(如叔丁基甲酸、1-金剛烷甲酸等)顯示出很好的催化活性(圖13)。
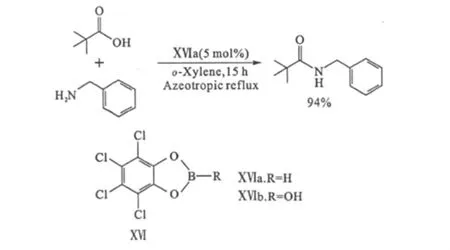
圖13 基于四氯代鄰苯二酚的催化劑ⅩⅥFig.13 CatalystⅩⅥ based on tetrachlorocatechol
ⅩⅥb可以通過四氯代鄰苯二酚和硼酸在直接酰胺化反應前現場制備。對于芳基硼酸類催化劑,往往制備上較為困難,限制了其應用,但ⅩⅥb制備十分簡便,且可以替代四氯代鄰苯二酚的二酚、二醇和二酸的結構非常多,應用前景廣闊。
此外,在早期的專利中還報道了其它硼化合物,如偏硼酸(HBO2)、氧化硼(B2O3)、三氟化硼-乙醚絡合物等也具有與硼酸相似的催化直接酰胺化反應的能力[16]。但是這些硼化合物對一般酰胺鍵形成的催化作用還有待進一步研究。
3 結語
從最初需要在回流條件下起作用的催化劑Ⅰ,到在室溫下就可以方便使用的催化劑Ⅹ和ⅩⅢ,硼化合物催化的直接酰胺化為酰胺的合成提供了全新途徑。該途徑具有原子經濟、過程簡便和成本低廉的特點,其中硼酸催化劑已可以應用到酰胺化合物的工業化大規模制備中。但對于更廣范圍的應用,還需要研究者進一步提升這些催化劑的活性,拓寬其適用的底物范圍,并使反應條件變得更加溫和;同時還需要對此類反應的機理進行深入的研究,以便能更好地指導催化劑的設計與優化。相信在不遠的未來,硼化合物催化的直接酰胺化能夠成為實驗室與工業上都廣泛采用的綠色、高效、簡便的酰胺合成方法。
[1]Roughley S D,Jordan A M.The medicinal chemist′s toolbox:An analysis of reactions used in the pursuit of drug candidates[J].J Med Chem,2011,54(10):3451-3479.
[2]March J,Smith M B.Advanced Organic Chemistry(6th ed)[M].New Jersey:John Wiley & Sons,2007:1841-1842.
[3]El-Faham A,Albericio F.Peptide coupling reagents,more than a letter soup[J].Chem Rev,2011,111(11):6557-6602.
[4]Montalbetti C A G N,Falque V.Amide bond formation and peptide coupling[J].Tetrahedron,2005,61(46):10827-10852.
[5]陸熙炎.綠色化學與有機合成及有機合成中的原子經濟性[J].化學進展,1998,10(2):123-130.
[6]Constable D J C,Dunn P J,Hayler J D,et al.Key green chemistry research areas—A perspective from pharmaceutical manufacturers[J].Green Chem,2007,9:411-420.
[7]Schfer G,Matthey C,Bode J W.Facile synthesis of sterically hindered and electron-deficient secondary amides from isocyanates[J].Angew Chem Int Ed,2012,51(36):9173-9175.
[8]Pattabiraman V R,Bode J W.Rethinking amide bond synthesis[J].Nature,2011,480:471-479.
[9]Charville H,Jackson D A,Hodges G,et al.The uncatalyzed direct amide formation reaction——Mechanism studies and the key role of carboxylic acid H-bonding[J].Eur J Org Chem,2011,2011(30):5981-5990.
[10]Mitchell J A,Reid E E.The preparation of aliphatic amides[J].J Am Chem Soc,1931,53(5):1879-1883.
[11]Webb C N.Benzanilide[J].Org Synth,1927,7:6-7.
[12]Lundberg H,Tinnis F,Adolfsson H.Direct amide coupling of nonactivated carboxylic acids and amines catalysed by zirconium(Ⅳ)chloride[J].Chem Eur J,2012,18(13):3822-3826.
[13]Comerford J W,Clark J H,Macquarrie D J,et al.Clean,reusable and low cost heterogeneous catalyst for amide synthesis[J].Chem Commun,2009,(18):2562-2564.
[14]Nelson P,Pelter A.Trisdialkylaminoboranes:New reagents for the synthesis of enamines and amides[J].J Chem Soc,1965:5142-5144.
[15]Sowa F J,Nieuwland J A.Organic reactions with boron fluoride.ⅩⅣ.The reaction of amides with acids and amines[J].J Am Chem Soc,1937,59(7):1202-1203.
[16]Ikeda T,Higuchi S,Hashimoto K,et al.Production of N-acylaminoacid amides[P].JP 61 000 050A,1986-01-06.
[17]Hall D G.Boronic Acids:Preparation and Applications in Organic Synthesis and Medicine[M].Weinheim:Wiley-VCH,2005:1-99.
[18]Ishihara K,Ohara S,Yamamoto H.3,4,5-Trifluorobenzeneboronic acid as an extremely active amidation catalyst[J].J Org Chem,1996,61(13):4196-4197.
[19]Ishihara K,Ohara S,Yamamoto H.(3,4,5-Trifluorophenyl)boronic acid-catalyzed amide formation from carboxylic acids and amines:N-benzyl-4-phenylbutyramide[J].Org Synth,2002,79:176-185.
[20]Ishihara K,Ohara S,Yamamoto H.Direct polycondensation of carboxylic acids and amines catalyzed by 3,4,5-trifluorophenylboronic acid[J].Macromolecules,2000,33(10):3511-3513.
[21]Ishihara K,Kondo S,Yamamoto H.3,5-Bis(perfluorodecyl)phenylboronic acid as an easily recyclable direct amide condensation catalyst[J].Synlett,2001,(9):1371-1374.
[22]Maki T,Ishihara K,Yamamoto H.N-Alkyl-4-boronopyridinium salts as thermally stable and reusable amide condensation catalysts[J].Org Lett,2005,7(22):5043-5046.
[23]Georgiou I,Ilyashenko G,Whiting A.Synthesis of aminoboronic acids and their applications in bifunctional catalysis[J].Acc Chem Res,2009,42(6):756-768.
[24]Arnold K,Davies B,Giles R L,et al.To catalyze or not to catalyze?Insight into direct amide bond formation from amines and carboxylic acids under thermal and catalyzed conditions[J].Adv Synth Catal,2006,348(7-8):813-820.
[25]Arnold K,Batsanov A S,Davies B,et al.Synthesis,evaluation and application of novel bifunctional N,N-diisopropylbenzylamine-boronic acid catalysts for direct amide formation between carboxylic acids and amines[J].Green Chem,2008,10(1):124-134.
[26]Arnold K,Davies B,Herault D,et al.Asymmetric direct amide synthesis by kinetic amine resolution:A chiral bifunctional aminoboronic acid catalyzed reaction between a racemic amine and an achiral carboxylic acid[J].Angew Chem Int Ed,2008,47(14):2673-2676.
[27]Al-Zoubi R M,Marion O,Hall D G.Direct and waste-free amidations and cycloadditions by organocatalytic activation of carboxylic acids at room temperature[J].Angew Chem Int Ed,2008,47(15):2876-2879.
[28]Gernigon N,Al-Zoubi R M,Hall D G.Direct amidation of carboxylic acids catalyzed by ortho-iodo arylboronic acids:Catalyst optimization,scope,and preliminary mechanistic study supporting apeculiar halogen acceleration effect[J].J Org Chem,2012,77(19):8386-8400.
[29]Marcelli T.Mechanistic insights into direct amide bond formation catalyzed by boronic acids:Halogens as Lewis bases[J].Angew Chem Int Ed,2010,49(38):6840-6843.
[30]Kondaiah G C M,Reddy L A,Babu K S,et al.Boric acid:An efficient and environmentally benign catalyst for transesterification of ethyl acetoacetate[J].Tetrahedron Lett,2008,49(1):106-109.
[31]Nguyen T B,Sorres J,Tran M Q,et al.Boric acid:A highly efficient catalyst for transamidation of carboxamides with amines[J].Org Lett,2012,14(12):3202-3205.
[32]Tang P.Boric acid catalyzed amide formation from carboxylic acids and amines:N-Benzyl-4-phenylbutyramide[J].Org Synth,2005,81:262-272.
[33]Mylavarapu R K,Kondaiah G C M,Kolla N,et al.Boric acid catalyzed amidation in the synthesis of active pharmaceutical ingredients[J].Org Proc Res Dev,2007,11(6):1065-1068.
[34]MaraN,Kocevar M.Boric acid-catalyzed direct condensation of carboxylic acids with benzene-1,2-diamine into benzimidazoles[J].Helv Chim Acta,2011,94(10):1860-1874.
[35]Maki T,Ishihara K,Yamamoto H.4,5,6,7-Tetrachlorobenzo[d][1,3,2]dioxaborol-2-ol as an effective catalyst for the amide condensation of sterically demanding carboxylic acids[J].Org Lett,2006,8(7):1431-1434.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
