Red同源重組在大腸桿菌基因敲除中的應用
2013-08-14 09:08:30呂沈聰趙穎穎鐘衛鴻
化學與生物工程 2013年6期
呂沈聰,趙穎穎,鐘衛鴻
(浙江工業大學生物與環境工程學院,浙江 杭州310032)
隨著多種細菌基因組測序工作的完成,獲得了大量的分子遺傳學信息。然而對分子遺傳結構以及其中編碼基因的了解卻很有限。基因突變提供了一種了解單個基因功能的方法。基因的修飾以及修飾后細菌表型的研究對于功能基因組學的發展尤為重要。
20世紀80年代以來,生物學家發展了一種新型分子生物學技術——基因敲除。至今已涌現出多種不同的基因敲除技術[1-3],廣泛應用于分子生物學、醫學細胞生物學、食品發酵工程等多個領域,展現出極大的發展空間。
大腸桿菌作為分子生物學的重要研究對象,是生產重組蛋白、氨基酸、有機酸等昂貴物質的主要微生物。通過修飾大腸桿菌的基因組,改變其代謝途徑,以發酵生產有用物質,已成為國內外研究的熱點。作者在此簡要介紹了近幾年發展的幾種不同的Red同源重組技術及其在大腸桿菌基因敲除中的應用。
1 Red同源重組作用機理
長的同源臂,往往長達幾百個堿基,而且同源重組效率低,往往不能得到所需的重組子。
1998年,Murphy[4]首次報道了利用λ噬菌體Red同源重組系統在大腸桿菌中進行基因修飾的方法,將λ噬菌體的重組功能基因exo、bet、gam克隆到質粒pTP223上,用于野生型大腸桿菌宿主的基因替換。Red同源重組又稱ET重組[5],它不依賴宿主本身的重組系統,編碼Exo、Beta、Gam 3種蛋白質。Exo蛋白是λ核酸外切酶,可以結合在雙鏈DNA的末端,從5′端向3′端降解 DNA 單鏈,產生3′突出末端[6]。bet基因編碼的Beta蛋白可以介導互補鏈之間的退火[7-10]。gam 基因編碼一個分子量為16 000Da的多肽,該多肽可與宿主RecBCD核酸外切酶結合,抑制核酸外切酶活性,防止外源DNA進入細胞后被宿主降解[11,12]。這3種蛋白協同作用,促進同源重組的發生。該方法以其較短的同源臂、較高的重組效率等特點廣泛應用于大腸桿菌的基因敲除中。
大腸桿菌基因敲除的傳統方法是利用其自身的RecA同源重組系統編碼的RecA和RecBCD蛋白介導DNA的同源重組。但是以該系統為基礎的基因打靶存在較多的缺點:首先,由于RecBCD具有核酸外切酶活性,線性的打靶DNA將被降解,打靶基因必須整合于載體上才能進行同源重組;其次,打靶片段需要較
2 幾種不同的Red同源重組技術
2.1 兩步同源重組法
2000年,Datsenko等[13]提出了兩步同源重組法在大腸桿菌K-12中進行基因敲除的策略。該方法以pKD46[14]作為同源重組質粒,該質粒為低拷貝溫敏型質粒,具有氨芐抗性,并將噬菌體的exo、bet、gam基因整合于阿拉伯糖啟動子控制下;質粒pKD3和pKD4為打靶DNA的獲得提供模板,分別帶有氯霉素和卡那霉素抗性基因,并賦有FRT位點[15](翻轉酶結合位點);pCP20質粒含有flippase基因[15],表達的 FLP翻轉酶可以與FRT位點結合并消除抗性基因片段。
方法:第一步,將一段含有FRT位點的抗性基因置換入宿主染色體待敲除區域;第二步,將質粒pCP20電轉化入宿主細胞,利用其表達的FLP翻轉酶將抗性基因敲除。
將pKD46轉化入宿主菌中,使其能夠表達λRed重組酶。以pKD3或pKD4為模板,PCR合成一段兩端與目的基因同源、中間含有抗性基因的雙鏈打靶DNA[16]。將該片段電轉化[17]入宿主細胞。L-阿拉伯糖誘導重組酶的表達,在重組蛋白的作用下,打靶DNA和細菌染色體基因的同源處發生交換,將抗性基因置換入宿主染色體,使其獲得相應抗性。復蘇后抗性平板篩選得到突變子。將質粒pCP20電轉化入宿主,利用其表達的FLP翻轉酶將染色體上的抗性基因敲除[14],得到重組框架正確的突變株,如圖1所示。
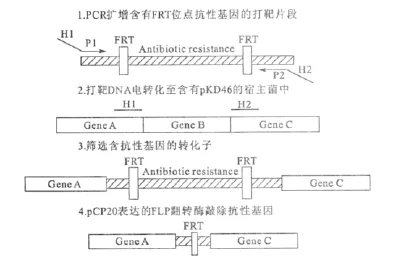
圖1 兩步法基因敲除步驟Fig.1 Procedure of two-steps gene knockout method
以敲除大腸桿菌 K-12的lac操縱子為例[13],敲除前后基因的序列結構如圖2所示。
分別以pKD3和pKD4為模板,PCR得到打靶DNA,將打靶片段電轉化入宿主細胞,在重組酶的作用下完成同源重組。抗性平板篩選轉化子。轉化子用各自位點特異性引物驗證,證明得到的轉化子獲得了預期大小的重組結構。測序結果表明lacZYA片段已經被準確敲除,留下完整的lacI片段。
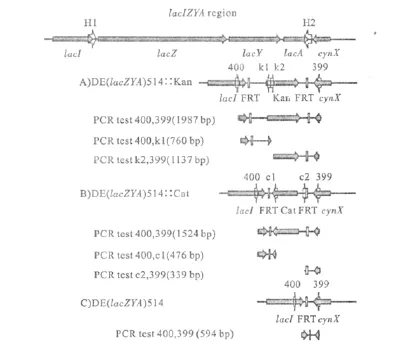
圖2 分別用pKD3和pKD4敲除大腸桿菌K-12 lac操縱子Fig.2 Gene knockout of K-12 lac operon using pKD3and pKD4,respectively
應用該敲除系統,美國普渡大學在大腸桿菌中成功敲除了40多個基因[13]。該方法所需要的打靶同源臂短,只需36~50bp,因此可以直接設計在引物上,而不需要DNA酶切和連接等程序,大大加快了實驗進程。
閥1處產生觸發指令時實際換相線電壓已經到達過零點,兩極電壓為正,閥1立刻導通,ca開始換相。同理,閥4處產生觸發指令時,為負,陰極電壓小于陽極電壓,立即導通,與上半橋一致。因此,當0<σca>σca
兩步同源重組法操作簡單、實驗周期短、打靶精確度高,是目前大腸桿菌基因敲除領域應用最為常見的方法。但該方法需要將外源DNA電轉化入宿主細胞,因此對DNA片段的濃度以及感受態的效率要求較高。然而某些菌株對外源DNA的吸收效率不高,進入細胞的DNA量往往不足以進行同源重組。同時,電轉化會導致大量細胞死亡,只有很少部分宿主細胞能存活[18]。導致兩步同源重組法在這些菌株中未能實現成功敲除。
2.2 雙質粒基因敲除法
雙質粒基因敲除法包含一個輔助質粒和一個供體質粒。供體質粒為打靶DNA提供來源,輔助質粒含有重組酶和歸巢內切酶基因。將兩種質粒共轉化入宿主細胞,在誘導劑的作用下,歸巢內切酶切割供體質粒產生打靶DNA,重組酶促進同源基因序列完成重組。
2.2.1 Gene gorging法
Gene gorging 法由 Herring 等[18]在 2003年報道,供體質粒為高拷貝質粒,常用的構建骨架為pUC19、pACYC184、pDHA30、pCR-BluntⅡ-Topo。通過酶切方法將打靶序列以及歸巢內切酶酶切位點(I-Sce I歸巢內切酶識別的長18bp的特異序列)克隆于質粒上。I-Sce I識別位點位于打靶序列兩側。輔助質粒pACBSR上整合了表達Red重組酶和I-Sce I內切酶的基因。在阿拉伯糖誘導下,表達的I-Sce I內切酶特異性切割供體質粒,產生打靶DNA。同時,Red重組酶則促進打靶DNA與宿主染色體同源序列之間的重組,實現基因敲除。
Gene gorging法不同于兩步同源重組法,其打靶片段不需要外源轉化,而是在宿主細胞內產生。質粒在宿主內大量復制,通過歸巢內切酶的切割,導致每個細胞都產生打靶DNA,數量大大增加,提高了可能發生同源重組的細胞基數。據報道,在不使用任何篩選標記的條件下,該方法篩選到突變株的概率達到1%~15%[18]。
某些大腸桿菌病原菌對于外源DNA的吸收效率低,運用兩步同源重組法很難得到重組子。Gene gorging法不依賴于外源DNA的轉化,適合于在這些菌株中進行基因敲除的嘗試。
Gene doctoring法包含供體質粒pDOC以及輔助質粒pACBSCE,原理與Gene gorging法類似,但在其基礎上增加了篩選條件,提高了篩選率。
供體質粒pDOC來源于pEX100T[19],整合了打靶基因的左右同源臂以及卡那霉素抗性基因,同源臂兩側為歸巢內切酶酶切位點,此外還攜帶sacB基因[19]。輔助質粒pACBSCE含有受阿拉伯糖調控的λ-Red重組酶基因和I-Sce I內切酶基因。在L-阿拉伯糖的誘導下,兩種酶在細胞內表達。I-Sce I內切酶切割pDOC產生線性DNA打靶片段,同時也切割pACBSCE,使該質粒逐漸消失,防止因λ-Red重組酶的過量表達導致染色體二次修飾[20-22]。在重組酶的作用下,線性打靶DNA和染色體的同源區發生交換,將卡那霉素抗性基因置換到宿主基因組上。菌液涂布于含有卡那霉素和蔗糖的LB瓊脂培養基上生長,篩選重組子。sacB基因的存在,使得含有供體質粒的菌株無法在含有蔗糖的培養基上存活,因為sacB基因表達的酶會水解蔗糖生成高分子果糖聚合物毒害宿主細胞。因此只有攜帶抗性基因、并且不含質粒pDOC的重組子才能在該培養基上生長,避免了假陽性轉化子的產生。FLP翻轉酶敲除篩選得到重組子的抗性片段,如圖3所示。
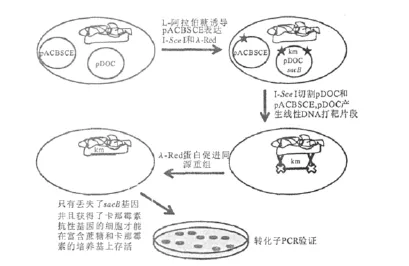
圖3 Gene doctoring實驗流程Fig.3 Procedure of gene doctoring experiment
Lee等將pDOC-C和pACBSCE分別電轉化入不同E.coli 菌 株 K-12MG1655[23]、EHEC O157:H7 Sakai和 UPEC CFT073[24]中,應用 Gene doctoring法對基因Rsd和Yocl進行敲除(表1)。
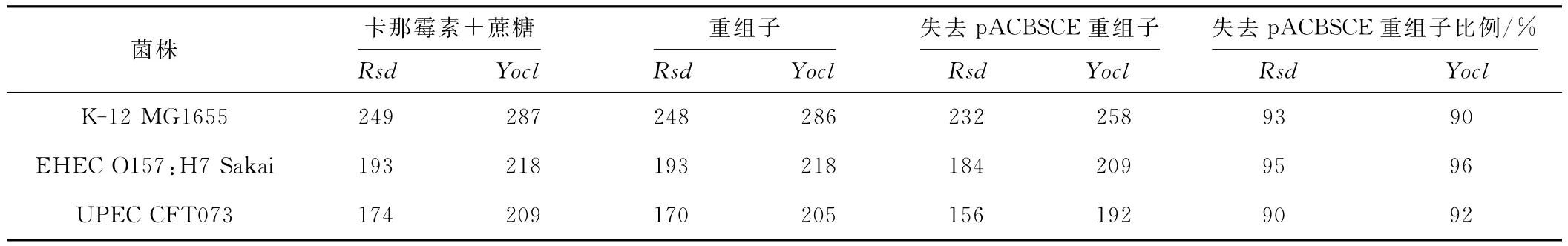
表1 Gene doctoring在不同E.coli菌株中重組效率的比較Tab.1 Comparison of recombination efficiency of different E.coli strains with gene doctoring method
在含有卡那霉素和蔗糖的培養基上長出大量轉化子。PCR驗證發現,絕大多數的克隆擁有正確的重組框架,并且超過90%的克隆對氨芐青霉素和氯霉素敏感,表明pDOC質粒和pACBSCE質粒已經在細胞中消除。其中一個克隆經PCR驗證,發現其待敲除部分基因框架與野生型的一樣,估計是卡那霉素基因插入到了該菌株染色體的其它部位。應用Gene doctoring法在這些菌株中進行敲除實驗,可以得到150個以上遺傳重組框架正確的突變子。
Lee等[19]比較了應用pACBSCE與 Gene gorging法中的pACBSR分別進行基因敲除的同源重組效率(表2)。
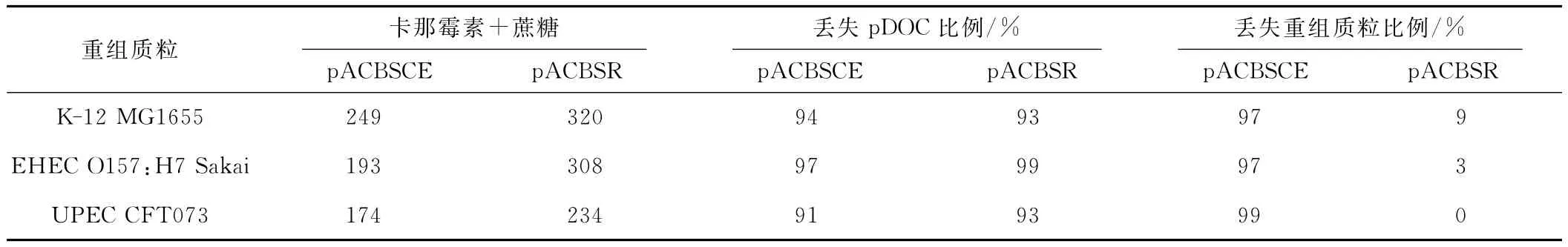
表2 應用質粒pACBSCE與質粒pACBSR同源重組效率的比較Tab.2 Comparison of recombination efficiency using recombineering plasmids pACBSCE and pACBSR
應用pACBSR作為重組質粒能夠得到更多具備卡那霉素抗性、對蔗糖不敏感的重組子,其中丟失pDOC質粒的克隆數跟應用pACBSCE質粒所得到的克隆數持平,但是其中大部分克隆仍含有pACBSR,只有很小部分消除了該重組質粒。這就導致宿主細胞會過多地暴露于重組蛋白的作用下而發生二次重組,需要 另 外 將 其 從 宿 主 中 消 除[10,25,26]。 因 此 選 用pACBSCE質粒更合適。
Gene doctoring法已經成功應用于各類大腸桿菌的基因敲除,包括一些運用兩步同源重組法很難敲除的致病菌。該方法不僅繼承了Gene gorging法高打靶效率的優點,并且增加了篩選標記,在完成基因敲除的同時消除了同源重組輔助質粒,提升了篩選效率。
2.3 單質粒敲除法
單質粒敲除法由Yu等[27]于2008年首次報道。該方法的特點在于只需單個質粒即可完成宿主基因組的修飾,并且在染色體上不留殘余序列,重組效率高。
質粒pREDI[27](圖4)攜帶有2個獨立的啟動子:一個為阿拉伯糖啟動子,可誘導λ-Red重組蛋白表達,介導帶標記的線性DNA打靶片段與目的基因區域的置換;另一個為鼠李糖啟動子,誘導I-Sce I歸巢內切酶的表達,切割基因組,促進雙鏈斷裂實現分子內重組。剔除篩選標記后,完成基因組的修飾。
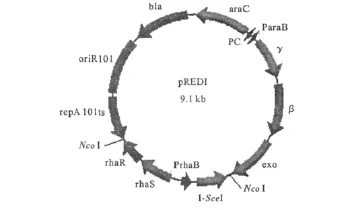
圖4 質粒pREDI圖譜Fig.4 The map of plasmid pREDI
圖5為應用質粒pREDI快速無痕敲除策略示意圖。為了敲除大腸桿菌基因組A區與C區之間的基因片段,通過PCR克隆出包含正篩選標記(CmR)、負篩選標記(sacB)、1個歸巢內切酶酶切位點和3段同源區(A-C)的線性DNA打靶片段。將線性DNA打靶片段電轉化入含有質粒pREDI的大腸桿菌中,涂布于含有阿拉伯糖的培養基上生長。在pREDI表達的重組酶作用下,打靶片段與目的基因發生置換。將菌株從含有10mmol·L-1阿拉伯糖的培養基轉移到含有10mmol·L-1鼠李糖的培養基中,誘導I-Sce I歸巢內切酶的表達。歸巢內切酶切割染色體上的I-SceI酶切位點,形成 DSB(雙鏈斷裂模型)[28-30]。DSB介導兩個同源區(boxC)之間的重組,去除外源整合片段,達到無痕修飾的目的。
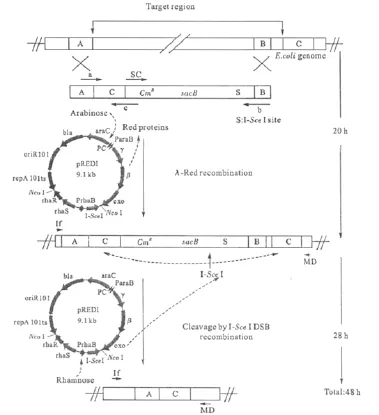
圖5 應用質粒pREDI快速無痕敲除策略Fig.5 Description of rapid markless gene knockout with plasmid pREDI
I-Sce I歸巢內切酶作用于細胞染色體上的酶切位點使DNA雙鏈斷裂,導致細胞死亡,因此只有去除了整合位點、修復了缺口、形成了正確突變結構的細胞才能存活。據報道,該方法的敲除成功率達到70%~100%[27]。
單質粒敲除法只需單個質粒pREDI就可成功進行基因組上的突變,完成一次敲除只需要2d,而且突變菌株染色體上不留任何痕跡。相比于之前的方法有了更大的進步,適用于一系列的基因修飾工作,例如在大腸桿菌或其它革蘭氏陰性菌株中進行基因的點突變、基因定點插入等。
3 結語
兩步同源重組法所需同源臂短,不需要構建于載體上,非常適合應用于常見大腸桿菌的基因敲除,例如K系列菌株,對外源DNA的吸收效率較高,打靶成功率大。Gene gorging法不含篩選標記,因此在某些情況下篩選工作較大、實驗周期長。基于Gene gorging的Gene doctoring法以高打靶率、高篩選率等優點逐漸應用于各類致病菌的基因敲除,例如EHEC O157:H7Sakai和UPEC CFT073等,它們對外DNA吸收效率低,運用兩步同源重組法很難成功。單質粒敲除法實驗周期短、打靶效率較高,也逐漸應用于各類大腸桿菌的基因修飾工作中。
作為與人類最為密切的微生物,大腸桿菌的代謝工程研究意義重大。通過修飾其基因組,使其獲得新的優良性狀和生產能力是目前國內外研究的熱點。Red同源重組技術的誕生為大腸桿菌染色體的修飾提供了強有力的工具,是基因工程領域的一場革命。而且該技術在其它細菌中成功進行基因敲除的報道也越來越多。作為一項高效基因打靶技術,Red同源重組技術的應用領域將會越來越廣,有望為基因功能的改良及后基因組時代功能研究作出巨大貢獻。
[1]Russell C B,Thaler D S,Dahlquist F W.Chromosomal transformation of Escherichia coli recD strains with linearized plasmids[J].Journal of Bacteriology,1989,171(5):2609-2013.
[2]Hamilton C M,Aldea M,Washburn B K,et al.New method for generating deletions and gene replacements in Escherichia coli[J].Journal of Bacteriology,1989,171(9):4617-4622.
[3]Link A J,Phillips D,Church G M.Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli:Application to open reading frame characterization[J].Journal of Bacteriology,1997,179(20):6228-6237.
[4]Murphy K C.Use of bacteriophageλrecombination functions to promote gene replacement in Escherichia coli[J].Journal of Bacteriology,1998,180(8):2063-2071.
[5]Muyrers J P P,Zhang Y M,Stewart A F.Techniques:Recombinogenic engineering-new options for cloning and manipulating DNA[J].Trends in Biochemical Sciences,2001,26(5):325-331.
[6]Poteete A R.What makes the bacteriophage lambda Red system useful for genetic engineering:Molecular mechanism and biological function[J].Fems Microbiology Letters,2001,201(1):9-14.
[7]Copeland N G,Jenkins N A,Court D L.Recombineering:A powerful new tool for mouse functional genomics[J].Nature Reviews Genetics,2001,2(10):769-779.
[8]Muniyappa K,Shaner S L,Tsang S S,et al.Mechanism of the concerted action of recA protein and helix-destabilizing proteins in homologous recombination[J].Proceedings of the National Academy of Sciences of the United States of America,1984,81(9):2757-2761.
[9]Takahashi N,Kobayashi I.Evidence for the double-strand break repair model of bacteriophage lambda recombination[J].Proceedings of the National Academy of Sciences of the United States of America,1990,87(7):2790-2794.
[10]Ellis H M,Yu D,DiTizio T,et al.High efficiency mutagenesis,repair,and engineering of chromosomal DNA using single-stranded oligonucleotides[J].Proceedings of the National Academy of Sciences of the United States of America,2001,98(12):6742-6746.
[11]Murphy K C.Lambda Gam protein inhibits the helicase and chistimulated recombination activities of Escherichia coli RecBCD enzyme[J].Journal of Bacteriology,1991,173(18):5808-5821.
[12]Warming S,Costantino N,Court D L,et al.Simple and highly efficient BAC recombineering using galKselection[J].Nucleic Acids Research,2005,33(4):e36.
[13]Datsenko K A,Wanner B L.One-step inactivation of chromosomal genes in Escherichia coli K-12using PCR products[J].Proceedings of the National Academy of Sciences of the United States of America,2000,97(12):6640-6645.
[14]Doublet B,Douard G,Targant H,et al.Antibiotic marker modifications of lambda Red and FLP helper plasmids,pKD46and pCP20,for inactivation of chromosomal genes using PCR products in multidrug-resistant strains[J].Journal of Microbiological Methods,2008,75(2):359-361.
[15]Broach J R,Hicks J B.Replication and recombination functions associated with the yeast plasmid,2μcircle[J].Cell,1980,21(2):501-508.
[16]Court D L,Sawitzke J A,Thomason L C.Genetic engineering using homologous recombination[J].Annual Review of Genetics,2002,36:361-388.
[17]Orkin S.Molecular-Cloning——A Laboratory Manual,2nd Edition -Sambrook J,Fritsch E F,Maniatis T[J].Nature,1990,343(6259):604-605.
[18]Herring C D,Glasner J D,Blattner F R.Gene replacement without selection:Regulated suppression of amber mutations in Escherichia coli[J].Gene,2003,311:153-163.
[19]Lee D J,Bingle L E H,Heurlier K,et al.Gene doctoring:A method for recombineering in laboratory and pathogenic Esche-richia coli strains[J].BMC Microbiology,2009,9:252.
[20]Hobman J L,Patel M D,Hidalgo-Arroyo G A,et al.Comparative genomic hybridization detects secondary chromosomal deletions in Escherichia coli K-12MG1655mutants and highlights instability in the flhDCregion[J].Journal of Bacteriology,2007,189(24):8786-8792.
[21]Poteete A R,Fenton A C,Nadkarni A.Chromosomal duplications and cointegrates generated by the bacteriophage lamdba Red system in Escherichia coli K-12[J].BMC Molecular Biology,2004,5:22.
[22]Murphy K C,Campellone K G.Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E.coli[J].BMC Molecular Biology,2003,4:11.
[23]Blattner F R,Plunkett G 3rd,Bloch C A,et al.The complete genome sequence of Escherichia coli K-12[J].Science,1997,277(5331):1453-1462.
[24]Welch R A,Burland V,Plunkett G,et al.Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli[J].Proceedings of the National Academy of Sciences of the United States of America,2002,99(26):17020-17024.
[25]Jamsai D,Orford M,Nefedov M,et al.Targeted modification of a human beta-globin locus BAC clone using GETrecombination and an I-Sce I counterselection cassette[J].Genomics,2003,82(1):68-77.
[26]Tischer B K,von Einem J,Kaufer B,et al.Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli[J].Biotechniques,2006,40(2):191-197.
[27]Yu B J,Kang K H,Lee J H,et al.Rapid and efficient construction of markerless deletions in the Escherichia coli genome[J].Nucleic Acids Research,2008,36(14):e84.
[28]Choulika A,Perrin A,Dujon B,et al.Induction of homologous recombination in mammalian chromosomes by using the I-Sce I system of Saccharomyces cerevisiae[J].Molecular and Cellular Biology,1995,15(4):1968-1973.
[29]Rong Y S,Titen S W,Xie H B,et al.Targeted mutagenesis by homologous recombination in D.melanogaster[J].Genes Development,2002,16(12):1568-1581.
[30]Schmidt-Puchta W,Orel N,Kyryk A,et al.Intrachromosomal homologous recombination in Arabidopsis thaliana[J].Methods in Molecular Biology,2004,262:25-34.
