多同位素內標氣相色譜-質譜法測定食品中4種氯丙醇含量
2013-03-06 02:31:49吳少明傅武勝傅海慶方勤美
食品科學 2013年18期
關鍵詞:標準
吳少明,傅武勝*,傅海慶,方勤美,華 娟
(1.福建省人獸共患病研究重點實驗室,福建省疾病預防控制中心,福建 福州 350001;2.福建省產品質量檢驗研究院,福建 福州 350002;3.福建農林大學金山學院,福建 福州 350002;4.福建省農業科學院生物技術研究所,福建 福州 350003;5.福建農林大學食品科學學院,福建 福州 350002)
多同位素內標氣相色譜-質譜法測定食品中4種氯丙醇含量
吳少明1,2,傅武勝1,*,傅海慶3,方勤美4,華 娟5
(1.福建省人獸共患病研究重點實驗室,福建省疾病預防控制中心,福建 福州 350001;2.福建省產品質量檢驗研究院,福建 福州 350002;3.福建農林大學金山學院,福建 福州 350002;4.福建省農業科學院生物技術研究所,福建 福州 350003;5.福建農林大學食品科學學院,福建 福州 350002)
采用多同位素內標法,結合七氟丁酰基咪唑(HFBI)衍生化的氣相色譜-質譜(GC-MS)聯用技術,建立了食品中4種氯丙醇含量的檢測方法。對比了不同來源HFBI衍生試劑的作用性能,對定量內標物和校正標準的選擇進行了比較。結果表明:僅用2種同位素內標(3-氯-1,2-丙二醇-d5(或2-氯-1,3-丙二醇-d5)和1,3-二氯-2-丙醇-d5)校正,同樣可達到準確測定食品中4種氯丙醇含量的要求;若采用3-氯-1,2-丙二醇(3-MCPD)為標準物定量測定2-氯-1,3-丙二醇(2-MCPD)時,所得2-MCPD結果偏低,僅為以2-MCPD為標準物定量結果(真實值)的52.1%~77.8%。在最佳條件下進行了方法學指標的系統驗證。4種氯丙醇的最低檢出限為2.5~10μg/kg;在100~2000ng范圍內,氯丙醇與內標物峰面積的比值和質量呈線性關系(R2>0.9990)。以醬油和方便面調料粉為基質,進行100、200、400μg/kg三個水平的加標回收實驗,4種氯丙醇的回收率均值為90.1%~107%,相對標準偏差(RSD)為3.37%~10.4%(n=6),均滿足痕量分析的要求。
食品;氯丙醇;基質分散固相萃取法;同位素稀釋技術;氣相色譜-質譜法
氯丙醇是某些食品中存在的一類化學污染物[1],主要包括單氯取代的3-氯-1,2-丙二醇(3-MCPD)與2-氯-1,3-丙二醇(2-MCPD)和雙氯取代的1,3-二氯-2-丙醇(1,3-DCP)與2,3-二氯-1-丙醇(2,3-DCP)。3-MCPD具有生殖毒性和腎臟毒性,為潛在致癌物,1,3-DCP為動物遺傳毒性致癌物[2],JECFA制定了3-MCPD的每日最大耐受劑量(PMTDI)為2μg/(kg bw?d)[3],尚無其他3種氯丙醇的PMTDI值。氯丙醇為酸水解植物蛋白(HVP)生產中容易形成的污染物[4],配制醬油、方便面調料粉等食品通常添加HVP以增加產品鮮味,由此造成這些食品氯丙醇的污染[5]。許多國家已對HVP、醬油等食品制定了氯丙醇的限量標準。2012年開始,我國規定固態、液態調味品(含配制醬油等) 3-MCPD須分別低于1.0、0.4mg/kg[6],歐盟甚至要求低于0.02mg/kg[7],這對氯丙醇的檢測提出了很高的要求。
目前的檢測方法主要針對氯丙醇中的3-MCPD和(或)1,3-DCP[8-19],國際公認的方法為穩定性同位素稀釋氣相色譜-質譜聯用法[4,8,10,15,19]。但食品中可能還伴隨著另外兩種氯丙醇(2-MCPD和2,3-DCP)的存在[20],由于2-MCPD與3-MCPD、1,3-DCP與2,3-DCP互為同分異構體,因此它們可能具有相似的毒性作用,這使得研究同時準確測定4種氯丙醇的方法成為必要。而目前僅見少量文獻[21-22]報道以d5-3-MCPD、d5-1,3-DCP為校正內標,同時測定醬油等食品中的4種氯丙醇。但氯丙醇的化學性質存在差異,僅用2種內標是否可準確測定4種氯丙醇,還需進一步驗證。此外,以3-MCPD為標準物測定2-MCPD的含量可能不準確[23]。因此,本實驗以4種氯丙醇各自對應的同位素(4種)為內標,建立同時測定食品中4種氯丙醇的方法,同時評估了內標物和標準物對測定結果準確性等指標的影響。
1 材料與方法
1.1 材料與試劑
釀造醬油和方便面調料粉購自福州當地超市。
3-氯-1,2-丙二醇(CAS號:96-24-2)、2,3-二氯-1-丙醇(CAS號:616-23-9)、3-氯-1,2-丙二醇-d5(同位素純度97%)、1,3-二氯-2-丙醇-d5(同位素純度98%)、2,3-二氯-1-丙醇-d5(同位素純度98%) 美國Aldrich公司;2-氯-1,3-丙二醇(CAS號:497-04-1)、2-氯-1,3-丙二醇-d5(同位素純度98%) 加拿大TRC公司;雙氯取代的1,3-二氯-2-丙醇(CAS號:96-23-1) 德國Fluka公司;氯化鈉、無水硫酸鈉(560℃烘烤6h后使用)、無水乙醚(經重蒸餾后使用)(均為分析純) 中國國藥集團上海化學試劑公司;硅藻土(編號29111) 美國Agilent公司;正己烷(色譜純) 美國Fisher公司;異辛烷(色譜純) 英國Pooled公司;七氟丁酰基咪唑(HFBI)購自Regis、TCI、Addams、Sigma、Alfa等公司以及實驗室合成;聚丙烯固相萃取柱(45mL/支)福州勤鵬生物技術公司。
1.2 儀器與設備
3800型氣相色譜儀和Saturn2000型質譜儀 美國Varian公司;DB-5MS毛細管柱(30m×0.25mm,0.25μm)美國Agilent公司;G-560E型漩渦混合器 美國Scientific Industries公司;KQ-500B型超聲波清洗器 昆山市超聲儀器公司;DHG-9123A型電熱恒溫鼓風干燥箱 上海精宏試驗設備公司;DFY40型搖擺式高速萬能粉碎機 溫嶺市林大機械公司;TM-0912P型馬弗爐 北京盈安美誠科學儀器公司;CP225D型電子天平(準確至0.01mg,用于稱量標準品) 德國Sartorius公司;YP202型電子天平 上海精密科學儀器公司;EYELA型旋轉蒸發儀 日本東京理化公司;0.5mL氣密針 美國Hamilton公司;移液器4把(最大量程分別為20、100、200μL和1000μL) 法國Gilson公司。
1.3 方法
1.3.1 標準溶液與標準曲線制備
標準儲備液:分別準確稱取0.5~1.0mg各氯丙醇及內標標準品于8支10mL容量瓶中,用乙酸乙酯溶解、定容至刻度線,充分混勻得到質量濃度為0.1mg/mL的標準和內標儲備液,于-24℃冰箱中放置。
標準中間液:取適量上述標準儲備液于同一支10mL容量瓶中,用正己烷稀釋至刻度,得到質量濃度為20μg/mL的混合標準中間液。按照同樣方法,配制混合內標中間液。
標準工作液:取適量上述標準中間液用正己烷稀釋,配制4.0?g/mL的混合標準工作液;同樣取上述內標中間液適量,以正己烷稀釋定容至10mL,得到4.0?g/mL的混合內標工作液。
標準系列:分別取上述標準工作液25、50、125、250、500μL于5支5mL玻璃試管中,并加入75μL混合內標工作液(4.0?g/mL),然后補加正己烷至體積約為1.0mL,制得氯丙醇質量濃度為100、200、500、1000、2000ng/mL的標準系列。此系列溶液經HFBI衍生后,以GC-MS進樣分析。
1.3.2 樣品前處理
1.3.2.1 加入內標
醬油等液體樣品:稱取3.0g樣品于50mL塑料離心管中,加入75μL混合內標溶液(4.0?g/mL),超聲5min,混勻后待凈化用。調料粉樣品:稱取1.0g高速粉碎均勻的樣品于15mL塑料離心管中,加入75μL混合內標溶液(4.0?g/mL),然后加入4.0g飽和氯化鈉溶液,超聲5min,使粉末充分分散于液體中,混勻后以5000r/min的轉速離心5min,將上層溶液轉移至50mL塑料離心管中,待凈化用。
1.3.2.2 基質分散固相萃取凈化
于上述溶液中加入3.0g硅藻土,拌勻后,將其全部轉入預先裝有1.5g硅藻土的固相萃取柱中。先以30mL正己烷淋洗,棄去淋洗液,再以80mL重蒸無水乙醚分3~4次加入萃取柱中,洗脫氯丙醇,流速不超過8mL/min,洗脫液以預先裝有約10g無水硫酸鈉的250mL三角瓶收集。洗脫結束后,每隔10min振搖三角瓶,以打散結塊的硫酸鈉,最大程度脫除洗脫液中的水分。
1.3.2.3 濃縮
將上述經充分脫水的洗脫液轉入250mL雞心瓶中,再以10mL正己烷洗滌無水硫酸鈉層,洗液一并轉入雞心瓶中。溶液減壓濃縮至約0.5mL,并小心將之轉移至5.0mL磨口玻璃刻度試管中,再用少量正己烷洗滌雞心瓶2~3次,洗液一并轉入試管中,至體積約為1.2mL,供衍生用。
1.3.2.4 衍生
于上述盛有濃縮液或標準液的刻度試管中,快速加入50μL HFBI衍生試劑,迅速蓋上磨口塞并渦旋混勻,于75℃恒溫烘箱中保溫15min后,逐一檢查每一試管,如發現試管內液體少于0.8mL,則補加正己烷至1.2mL,并在75℃條件下繼續加熱15min。反應結束后,取出試管,待其冷卻至室溫后,用異辛烷定容至1.0mL,并加入2mL飽和氯化鈉溶液,充分渦旋,使得下層液體澄清。靜置后,取上層液體于事先裝有約0.3g無水硫酸鈉的2mL進樣瓶中,供GC-MS分析。
1.3.3 GC-MS分析條件
氣相色譜條件:DB-5MS毛細管柱(30m×0.25mm,0.25μm);進樣口溫度:280℃;程序升溫條件:50℃保持1min,再以2℃/min的速度升至78℃,最后以30℃/min的速度升至300℃,保持5min;載氣:高純氦,流速0.8mL/min;進樣方式:不分流進樣;進樣體積:1μL。質譜條件:電子電離(electron ionization,EI)源;電子倍增器增益:+250V;燈絲電流:100μA;阱溫:230℃;傳輸線溫度:250℃;歧盒溫度:50℃;溶劑延遲:3min;質譜采集時間:3~29min;掃描質量數范圍:m/z 90~300;掃描速率:0.65s/次。
2 結果與分析
2.1 定量離子的選擇
將質量為5000ng的氯丙醇及其同位素內標物標準液單獨衍生,對衍生物進行全掃描。3-MCPD、2-MCPD的m/z 253的靈敏度最高,m/z 289、m/z 453次之,文獻大多采用m/z 253定量[10,13,15-17]3-MCPD,但在本體系中,以m/z 253得到的提取離子色譜圖,在待測目標物處有明顯的干擾峰,因此選擇m/z 289作為3-MCPD、2-MCPD的定量離子;d5-3-MCPD與d5-2-MCPD均采用m/z 257;1,3-DCP采用m/z (275+277)靈敏度明顯優于文獻[10,13,15-17]的m/z 275定量,對于其同位素內標d5-1,3-DCP,則采用m/z (278+280)定量;2,3-DCP以m/z (75+77)同樣優于文獻[15]的m/z 75,d5-2,3-DCP則采用m/z 79。表1列舉了氯丙醇及其內標的監測離子和定量離子,提取離子色譜圖見圖1。
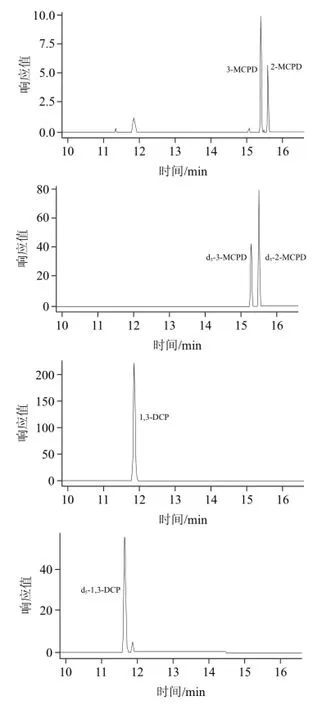
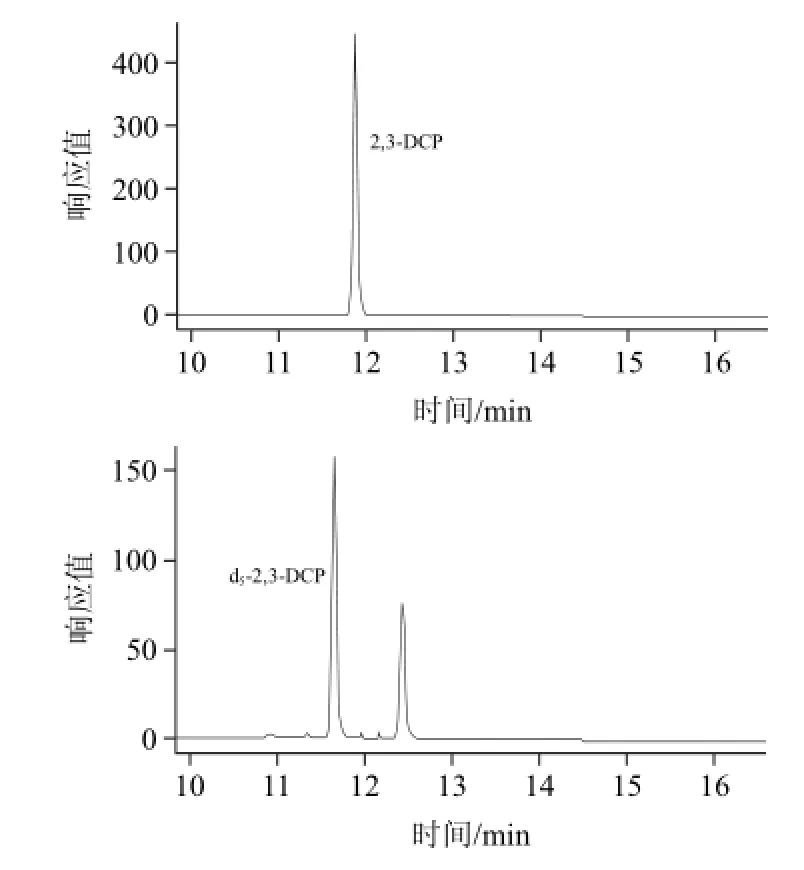
圖1 氯丙醇及其內標衍生物提取離子色譜圖Fig.1 Extracted ion chromatograms of the derivatives of chloropropanols and internal standards
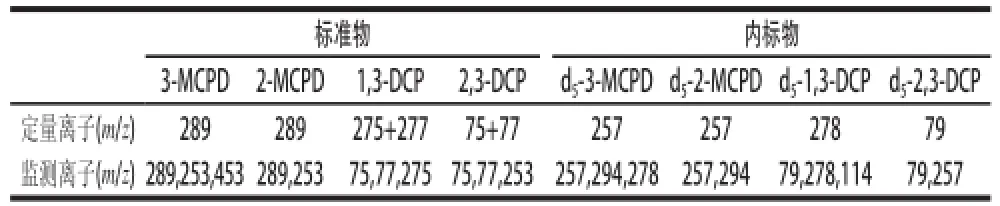
表1 氯丙醇及其內標衍生物的監測離子和定量離子Table1 Quantitation and monitoring ions for the derivatives of chloropropanols and internal standards
2.2 HFBI衍生試劑性能對比
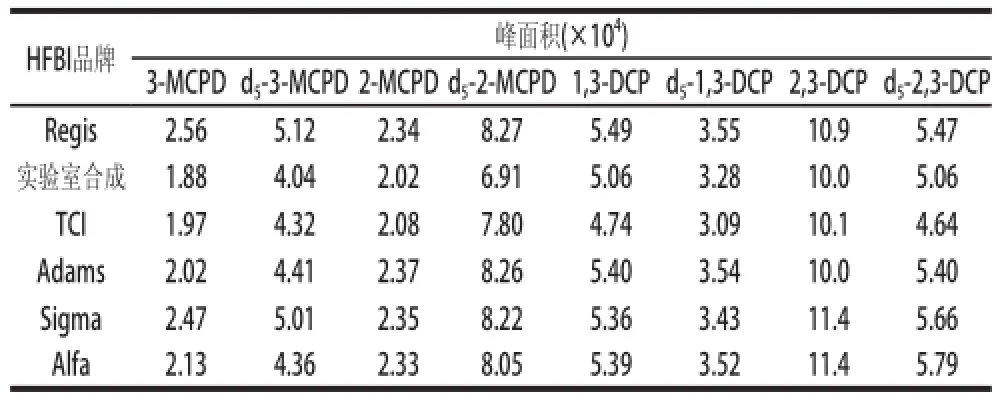
表2 不同品牌HFBI衍生后氯丙醇峰面積的對比((n=3)Table2 Comparison of the peak areas of chloropropanols and internal standards after derivatized by different brands of HFBI (
以醬油為基質,加入氯丙醇混合標準液和對應的內標混合液各600ng,按1.3.2節進行預處理,洗脫液濃縮至約7mL。準確移取6份于編號的5mL刻度試管中,每份體積為1.0mL,分別加入50μL不同品牌的HFBI衍生試劑,衍生液用GC-MS進樣分析,每個條件重復實驗3次,結果見表2。對比6種品牌HFBI的衍生作用,同一目標物質的峰面積(響應)相差不大,3次重復測定結果的相對標準偏差(RSD)在1.1%~6.1%之間,但實驗室合成HFBI產品與TCI公司HFBI衍生時目標物響應稍低。比較不同衍生劑,對于每一氯丙醇,其與對應內標峰面積的比值均較為接近,RSD為1.2%~2.2%。Regis公司產品衍生后氯丙醇峰形最好,而實驗室合成衍生劑產品氯丙醇峰形略差。從包裝上看,Regis公司產品瓶口采用橡膠塞密封,更有利于氣密針穿刺取液,而且潮氣不易進入試劑瓶內,使衍生操作更簡便,且該公司產品價格也較低,本實驗選用Regis公司HFBI試劑。
2.3 同位素內標的使用和選擇
2.3.1 同位素內標的使用
將氯丙醇標準及其同位素內標加入到20% NaCl溶液中,按照1.3.2節樣品前處理步驟進行操作。若采用外標法計算氯丙醇的結果,并與以正己烷為介質的相同質量的標準液衍生物進行比較,4種氯丙醇的絕對回收率僅為35.2%~59.1%;而采用內標法計算時,其相對回收率則為91.2%~106%,滿足測定要求。此外,在對醬油以及方便面調料粉進行加標實驗時,單氯丙醇及其內標的絕對回收率僅為38.1%~56.5%,而雙氯丙醇及其內標的回收率更低,僅為6.34%~15.3%。由此可見,采用本實驗操作步驟,必須使用內標法才可獲得準確可靠的結果。
2.3.2 同位素內標的選擇
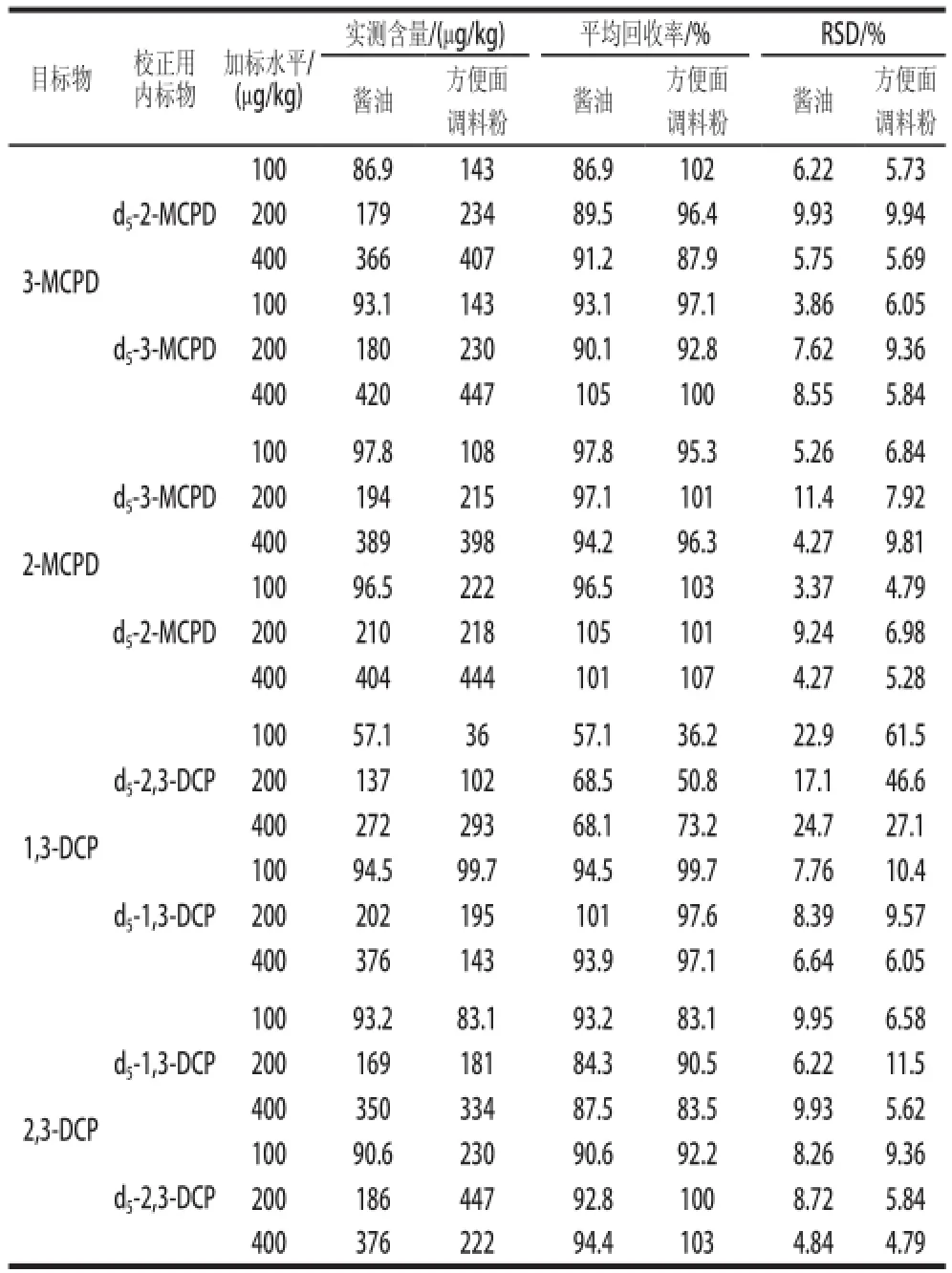
表3 內標物對測定結果的影響((n=6)Table3 Effect of internal standards on analysis results (n==66))
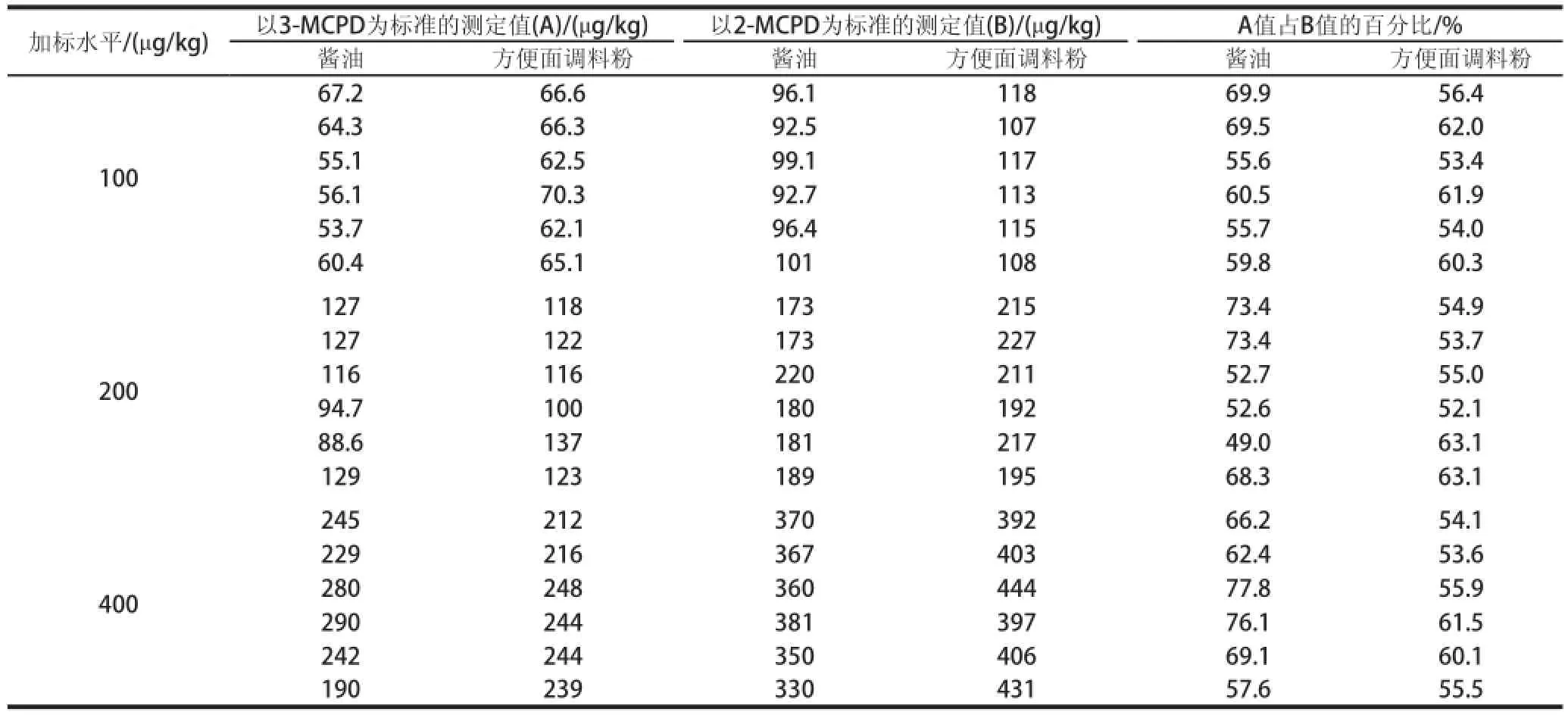
表4 定量標準對加標醬油和方便面調料粉樣品中2-MCPD含量的影響Table4 Effect of calibration standard of 3-MCPD and 2-MCPD on 2-MCPD content in spiked soy sauce and instant noodle seasoning powder
之前,由于國際上缺乏2-MCPD標準品和d5-2-MCPD和d5-2,3-DCP內標標準品出售,因此文獻[19,21-22]方法僅以d5-3-MCPD和d5-1,3-DCP為內標物校正3~4種氯丙醇,因此所測得結果可能不夠準確,需要對此予以重新評估。本實驗以市售釀造醬油和方便面調料粉為基質,進行3種水平(100、200μg/kg和400μg/kg)的加標回收實驗,結果見表3。當以d5-3-MCPD或d5-2-MCPD作為單氯丙醇的定量內標時,單氯丙醇的回收率為86.9%~107%,RSD為3.37%~11.4%,皆滿足痕量分析的要求。這說明d5-3-MCPD或d5-2-MCPD皆可作為單氯丙醇(3-MCPD和2-MCPD)測定的定量內標。以d5-1,3-DCP作為雙氯丙醇的定量內標時,目標物(1,3-DCP和2,3-DCP)的回收率均較為理想,在83.1%~101%之間,RSD為5.62%~11.5%。以d5-2,3-DCP作為雙氯丙醇的定量內標時,2,3-DCP的回收率和RSD分別為90.6%~103%、4.79%~9.36%,滿足要求;而1,3-DCP的回收率僅為36.2%~73.2%,RSD高達17.1%~61.5%。這說明僅d5-1,3-DCP可作為2種雙氯丙醇測定的定量內標,而d5-2,3-DCP作為1,3-DCP的定量內標時,所得結果不滿足要求。
2.3.3 2-MCPD測定時標準物的對比
之前,由于國際上缺乏商品2-MCPD標準品出售,文獻[22]報道以3-MCPD的標準曲線計算2-MCPD的含量。由于不同物質的響應不同,以此方法所得2-MCPD含量可能不夠準確。因此需要對該法的可靠性重新進行驗證。在上述醬油和方便面調料粉氯丙醇加標回收實驗中,采用2-MCPD、3-MCPD兩條標準曲線計算、對比2-MCPD的含量,并以2-MCPD為標準曲線測定結果作為真值進行評價,結果見表4。以3-MCPD標準曲線計算所得2-MCPD結果均偏低,釀造醬油、方便面調料粉中,以3-MCPD為標準曲線計算所得2-MPCD含量分別為真值的52.6%~77.8%、52.1%~63.1%,均不滿足測定的準確度要求。因此為了精確定量,必須以2-MCPD為標準物計算2-MCPD的含量。
2.4 方法學指標的考察
2.4.1 檢出限和定量限
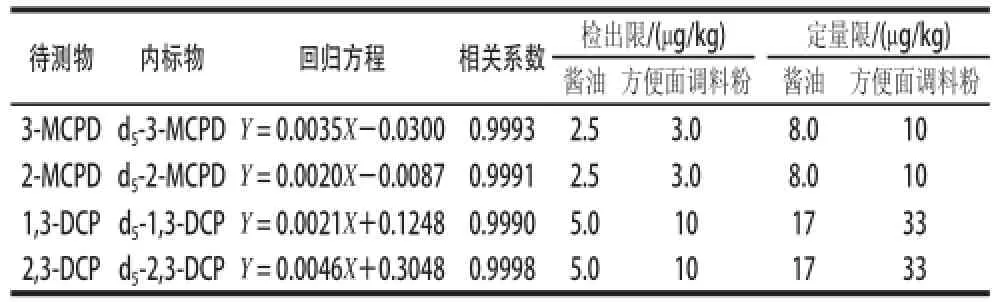
表5 氯丙醇的檢出限和定量限、線性回歸方程Table5 Regression equation, and detection and quantification limits of chloropropanols
如表5所示,在醬油基質與方便面調料粉基質中,4種氯丙醇的檢出限(LOD,RSN=3)在2.5~10μg/kg之間,定量限(LOQ,RSN=10)為8.0~33μg/kg。由于雙氯取代氯丙醇極性較單氯取代氯丙醇更小,在基質分散固相萃取(SPE)中,部分被正己烷淋洗造成靈敏度損失,因此其LOD和LOQ為單氯取代氯丙醇的2~3倍,但均滿足食品中痕量氯丙醇的分析要求。
2.4.2 線性范圍
以氯丙醇與各自內標物峰面積的比值Y和氯丙醇質量X(ng)計算線性回歸方程。在最優條件下進行方法學驗證實驗,在100~2000ng范圍線性關系良好,相關系數R均大于0.9990(表5),滿足分析要求。
2.4.3 精密度和準確度
以醬油和方便面調料粉(12種常見品牌方便面調料粉混勻)為基質,在最優條件下進行100、200、400μg/kg三個水平的加標回收實驗,每個水平重復6次。結果如表3所示,分別以各自對應的同位素內標校正時,4種氯丙醇的回收率均為90.1%~107%,RSD為3.37%~10.4%,重復性(精密度)和準確度均較為理想,滿足痕量分析的要求。
2.5 實際樣品分析
利用本方法調查了市售8種品牌方便面調料粉和7種品牌醬油氯丙醇的含量。結果表明,均未檢出雙氯丙醇(小于LOD);方便面調料粉和醬油樣品各有一份檢出單氯丙醇,方便面調料粉中3-MCPD、2-MCPD含量分別為0.97、0.05mg/kg,醬油中3-MCPD、2-MCPD含量分別為1.3、0.11mg/kg。3-MCPD含量超過我國限量要求(固態和液態分別為1.0、0.4mg/kg),均高于歐盟的限量值(0.02mg/kg)。
3 結 論
采用多同位素內標法,結合基質分散固相萃取法(MSPD),以氣相色譜-質譜法測定,建立了同時測定醬油和方便面調料粉中4種氯丙醇含量的方法。該方法明顯較GB/T 5009.191—2006《食品中氯丙醇含量的測定》法簡便,減少了取樣量和硅藻土、有機溶劑的消耗,靈敏度基本一致,精密度和準確性得到了進一步提高,并將該方法成功應用于實際樣品的分析。結果表明僅需2種內標(d5-1,3DCP和d5-3-MCPD或d5-2-MCPD)即可達到食品中4種氯丙醇準確定量的目的。同時若以3-MCPD標準曲線定量2-MCPD值時,2-MCPD僅為真實值的52.1%~77.8%。
[1] SCHALLSCHMIDT K, HITZEL A, POHLMANN M, et al. Determination of 3-MCPD in grilled meat using pressurized liquid extraction and gas chromatography-high resolution mass spectrometry[J]. Journal für Verbraucherschutz und Lebensmittelsicherheit, 2012, 7(3): 203-210.
[2] 嚴小波. 食品中脂肪酸氯丙醇酯和N-亞硝胺檢測方法的研究[D]. 福州: 福州大學, 2012.
[3] Scientific Committee on Food. Opinion of the scientific committee on food 3-monochloropropane-1,2-diol (3-MCPD)[EB/OL]. [2013-06-11]. www. europea. eu. int/comm. /food/fs/sc/scf/out91_en. pdf.
[4] 周相娟, 謝精精, 趙玉琪, 等. 氣相色譜-質譜法測定醬油中氯丙醇類化合物[J]. 中國調味品, 2011, 36(5): 88-90.
[5] 傅武勝, 吳永寧, 趙云峰. 調味品氯丙醇污染狀況與各國的危險性管理[J]. 中國調味品, 2003, 8(3): 14-19.
[6] GB 2762—2012 食品中污染物限量[S].
[7] Commission E. Commission Regulation (EC) No 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs[J]. Off J Eur Union L, 2006, 364(5): 1324-1328.
[8] RACAMONDE I, GONALEZ P, LORENZO R, et al. Determination of chloropropanols in foods by one-step extraction and derivatization using pressurized liquid extraction and gas chromatography-mass spectrometry[J]. Journal of Chromatography A, 2011, 1218(39): 6878-6883.
[9] LEON N, YUSA V, PARDO O, et al. Determination of 3-MCPD by GC-MS/MS with PTV-LV injector used for a survey of Spanish foodstuffs[J]. Talanta, 2008, 75(3): 824-831.
[10] ABUEIHAJ S, BOGUSZ J, IBRAHIM Z, et al. Rapid and simple determination of chloropropanols (3-MCPD and 1,3-DCP) in food products using isotope dilution GC-MS[J]. Food Control, 2007, 18(1): 81-90.
[11] 申中蘭, 郗丹, 盛建偉, 等. 固相萃取-氣相色譜-串聯質譜法(GCQqQ-MS/MS)測定醬油中三氯丙醇方法研究[J]. 齊魯師范學院學報, 2012, 27(5): 73-76.
[12] KISSA E. Determination of 3-chloropropanediol and related dioxolanes by gas chromatography[J]. Journal of Chromatography A, 2008, 605(1): 134-138.
[13] DAYRIT F M, NINONUEVO M R. Development of an analytical method for 3-monochloropropane-1,2-diol in soy sauce using 4-heptanone as derivatizing agent[J]. Food Additives and Contaminants, 2004, 21(3): 204-209.
[14] GB/T 5009. 191—2006 食品中氯丙醇含量的測定[S].
[15] HASNIP S, CREWS C, POTTER N, et al. Determination of 1, 3-dichloropropanol in soy sauce and related products by headspace gas chromatography with mass spectrometric detection: interlaboratory study[J]. Journal of AOAC International, 2010, 88(5): 1404-1412.
[16] CHUNG S W, KWONG K, YAU J C, et al. Chloropropanols levels in foodstuffs marketed in Hong Kong[J]. Journal of Food Composition and Analysis, 2008, 21(7): 569-573.
[17] GONZALEZ P, RACAMONDE I, CARRO A M, et al. Combined solid-phase extraction and gas chromatography-mass spectrometry used for determination of chloropropanols in water[J]. Journal of Separation Science, 2011, 34(19): 2697-2704.
[18] 馮笑軍. GC/MS 測定醬油中 3-氯丙醇含量的不確定度評定[J]. 廣州化工, 2012, 40(19): 92-94.
[19] 于杰, 李曉玉, 隋濤. GC-MS 快速檢測醬油中的氯丙醇[J]. 廣州化工, 2011, 39(11): 106-136.
[20] 傅武勝.食品中氯丙醇的檢測技術與暴露評估研究[D]. 北京: 中國疾病預防控制中心, 2005.
[21] XU X M, WU H W, HE H L, et al. Study of chloropropanols in soy sauce by gas chromatography-triple quadrupole mass spectrometry with coupled column separation without derivatization[J]. Food Additives and Contaminants, 2012, 30(3): 421-429.
[22] 傅武勝, 吳永寧, 趙云峰, 等. 穩定性同位素稀釋技術結合GC-MS測定醬油中多組分氯丙醇的研究[J]. 中國食品衛生雜志, 2004, 16(4): 289-294.
[23] 傅武勝, 趙云峰, 李敬光, 等. 我國市售水解蛋白氯丙醇污染狀況研究[J]. 食品科學, 2007, 28(1): 282-285.
Determination of 4 Kinds of Chloropropanols in Foods by Gas Chromatography-Mass Spectroscopy Coupled with Multi-Isotopic Internal Standard Technique
WU Shao-ming1,2,FU Wu-sheng1,*,FU Hai-qing3,FANG Qin-mei4,HUA Juan5
(1. Fujian Provincial Key Laboratory of Zoonosis Research, Center for Disease Control and Prevention, Fuzhou 350001, China;2. Fujian Inspection and Research Institute for Product Quality, Fuzhou 350002, China;3. College of Jinshan, Fujian Agriculture and Forestry University, Fuzhou 350002, China;4. Biotechnology Research Institute, Fujian Academy of Agricultural Sciences, Fuzhou 350003, China;5. College of Food Science, Fujian Agriculture and Forestry University, Fuzhou 350002, China)
An analytical method was developed to determine 4 kinds of chloropropanols in foods using gas chromatography mass spectrometer coupled with the multi-isotopic internal standard technique. The performance difference between different brands of HFBI as a derivativization reagent and internal standard selection for quantification were discussed. Under the optimal conditions, the limits of detection (LOD) were 2.5–10 μg/kg for 4 kinds of chloropropanols. According to the internal standard calibration, an excellent linearity between 100 ng and 2000 ng of chloropropanols (R2 > 0.9990) was obtained. When chloropropanols were spiked at levels of 100, 200, 400 mg/kg in soy sauce and seasoning powder of instant noodles, the average recovery rates were 90.1%–107%, with relative standard deviation (RSD) of 3.37%–10.4% (n = 6). The application of only 2 kinds of internal standards (chloropropane-1,2-diol-d5or 2-chloropropane-1,3-diol-d5and 1,3-dichloro-2-propanol-d5) can reveal accurate and precise determination for 4 kinds of chloropropanols. When 3-chloropropane-1,2-diol (3-MCPD) was used to calibrate 2-MCPD in foods, the level of 2-chloropropane-1,3-diol (2-MCPD) was only 52.1%–77.8% of the real content.
O657
A
1002-6630(2013)18-0131-06
10.7506/spkx1002-6630-201318026
2013-06-11
國家自然科學基金面上項目(81172671);衛生部公益性行業科研專項(200902009);福建省杰出青年基金項目(2011J06011);福建省醫學創新課題(2011-CX-21)
吳少明(1986—),男,檢驗員,碩士,研究方向為食品檢測。E-mail:280257959@qq.com
*通信作者:傅武勝(1971—),男,主任技師、副教授,博士,研究方向為污染物化學與食品安全。E-mail:fwsfqm@126.com
Key words:food;chloropropanol;dispersion matrix solid-phase extraction;isotopic internal standard technique;gas chromatography-mass spectrometer (GC-MS)
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
當代陜西(2019年8期)2019-05-09 02:22:48
上海建材(2019年1期)2019-04-25 06:30:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
質量與標準化(2015年9期)2015-12-31 11:41:40
中國質量與標準導報(2014年4期)2014-03-11 19:54:25
中國質量與標準導報(2014年10期)2014-02-28 22:25:47
中國質量與標準導報(2014年7期)2014-02-28 22:24:39
