新型染料木素磺酸酯的前藥判定與大鼠體內(nèi)生物利用度
2012-12-22 09:23:58劉新強(qiáng)鄧澤元
天然產(chǎn)物研究與開(kāi)發(fā) 2012年1期
彭 游,劉新強(qiáng),鄧澤元
1九江學(xué)院化工學(xué)院,九江332005;2南昌大學(xué)食品科學(xué)與技術(shù)國(guó)家重點(diǎn)實(shí)驗(yàn)室,南昌330047
Introduction
Soybean isoflavone is a class of secondary metabolite in soybean.In recent years the abundant researches indicated the soybean isoflavones in foods had many important physiological functions including preventing cancer,cardiovascular disease and the osteoporosis sickness,anti-oxidation,reducing the female menopause syndromea and the blood sugar,anti-senilly,etc[1-4].However,among the soybean isoflavones,GE includes three polar groups hydroxyl,its lipophilicity is weak.Simultaneously its hydroxyls formed the intermolecular hydrogen bond,its lattice energy is high,so its hydrophility is also weak.Only a small part of GE can be absorbed rapidly and metabolized to glucuronic acid glycosides in vivo,most were degraded and metabolized by microorganisms in the intestine.So,it has the first pass metabolism in the intestine[5].Thus,the defect of solubility and the first pass effect causes its low bioavailability and biological activity.Until now it cannot be used widespread effectively in clinical[6].To improve the oral bioavailability and pharmacological action of GE,our laboratory had designed and synthesized GB by the prodrug principle[7].We hope that com-pound can radically improve solubilities and metabolic stabilities,increase its in vivo bioavailability and anticancer activity(Fig.1).To isolate new prodrug that has a good affinity and high bioavailability,metabolism and pharmacokinetics of GB was studied in animals.If the original drug can be determined in plasma,the derivative is prodrug of GE.The pharmacokinetics of this prodrug corroborated was studied in vivo,to investigate whether its bioavailability was improved or not.Taking daidzein(DZ)as the internal standard,simultaneous determinations of GE and GB in biological samples were found by the HPLC method,and using the method to study in vivo metabolism and pharmacokinetics of GE and GB.
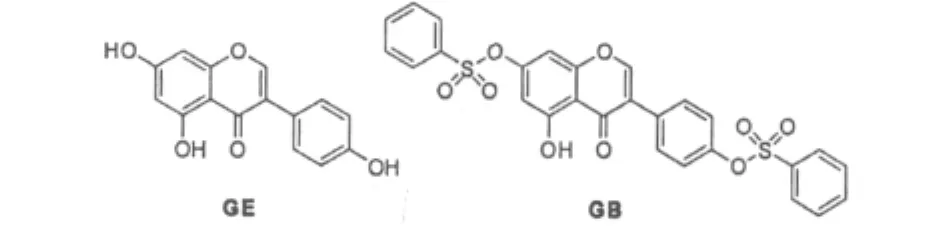
Fig.1 Structure of GE and its derivative
Materials and Methods
Reagents and Materials
Chemicals GB was synthesised by our lab(the purity,bigger than 99%,HPLC determination)[7].DZ and GE were provided by Shanxi Huike Botanical Development Co.,Ltd.HPLC-grade methanol(produced by Merck Corp.)were used.The other chemicals were of analytical grade.
Animals Fifteen of Wistar rats with female/male(2/3) at 200-250 g of body weight,were provided by the ExperimentalAnimalCenterofNanchangUniversity (Certificate of Conformity:SCXK-2009-0009).
Assay
Concentrations of drugs were determined in plasma using a RP-HPLC method.Analysis was performed on an Agilent 1100 HPLC system(Agilent Technologies,Palo Alto,CA,USA)equipped with a quaternary pump,a vacuum degasser,an automatic injector,and a variable wavelength detector.Separation was carried on a 5 μL Venusil XBP-C18column(250 mm ×4.6 mm,Agela Technologies,China)using 0.1%formic acid:methanol(1∶1,v/v)as the mobile phase at a flow rate of 0.5 mL/min.The effluent was monitored at 262 nm with detector sensitivity of 2.00 AUFS,and the maximum absorbance waves for GB was determined at 262 nm with a DAD detector.Agilent Chromatographic Station was used.The column temperature was kept at 20℃.The injected volume of the analyzed samples was 10 μL.
Plasma sample pretreatment
Following addition of 10 μL DZ solution(100 μg/mL) to 40 μL of plasma sample,the sample was swirled for 3 min.And then,1.0 mL of ethyl acetate were added to this sample.The sample was swirled for 3 min again and centrifuged at 14000 r/min for 10 min to separate layers at 4℃.The organic layer was transferred to clean tube and evaporated to dryness under N2.The residue was dissolved in 200 μL of methanol,and 10 μL was injected into the HPLC system.
Solution preparation
Administration of solution in drug metabolism
Taking minutely 50 mg of drug,added 0.1-0.2 mL dimethyl sulfoxide(DMSO),dissolved drug with ultrasound,diluted to 5.0 mL with PEG-400,so a concentration of 10 mg/mL solution of the drug was prepared that was used for oral administration of rats in the prodrug screening experiments.The solution was prepared just before use.
Administration of solution in pharmacokinetics
Drug solution of intragastric administration:Taking minutely 40 mg of GE(GB),added 0.1-0.2 mL DMSO,dissolved drug with ultrasound,diluted to 8.0 mL with PEG-400,so a concentration of 5 mg/mL solution of the drug was prepared that was used for oral administration of rats.The solution is prepared and used presently.
Prodrug solution of intravenous administration:Taking minutely 40 mg of GB,added 0.1-0.2 mL DMSO,dissolved drug with ultrasound,diluted to 2.0 mL with PEG-400,so a 20 mg/mL solution of the prodrug was prepared that was used for intravenous administration of rats.The solution was prepared just before use.
The drug metabolism research in vivo
A healthy Wistar rat with weight 200-250 g was fasted 12 h and given access to water freely before administra-tion.The drug GB was gavaged by 100 mg/kg dose to a rat.At 5,10,30 min and 1 h after administration,0.1-0.2 mL of blood were collected through the orbital venous plexus respectively.After centrifuged chilled 10 min with 14000 r/min,blood was separated to plasma.Combining of 0-1 h plasma samples,0.5 mL of the mixed sample were added 50 μL of methanol.The rear operation accorded“1.3 Plasma sample pretreatment”method.
The pharmacokinetics in rats
Dosage regimen in pharmacokinetics experiments and sample collection
Intragastric administration:Ten healthy Wistar rats,female/male(2/3),weight 200~250 g,were randomly divided into 2 treatment groups.All animals were fasted 12 h and accessed to water freely before administration.The drugs were gavaged by 40 mg/kg dose(dose volume of 10 mL/kg)respectively.At before administration(0 h),0.08,0.17,0.33,0.67,1,2,4,6,8,12,24 h after administration,0.1mL of blood were collected through the orbital venous plexus to heparin tube respectively.After centrifuged chilled at 14000 r/min for 10 min,the upper layer plasma sample was preserved at the-70℃ until analysis.
Intravenous medication:Randomly five healthy Wistar rats,female/male(2/3),weight 200-250 g,were divided into a treatment group.All animals were fasted 12 h and accessed to water freely before administration.Respectively the prodrug was injected via the tail vein by 40 mg/kg dose(dose volume of 2 mL/kg).At before administration(0 h),0.08,0.17,0.33,0.67,1, 2,4,6,8,12,24 h after administration,0.1 mL of blood were collected through the orbital venous plexus to heparin tube respectively.After centrifuged chilled at 14000 r/min for 10 min,the upper layer plasma sample were preserved at the-70℃ until analysis.
Determination of drug concentration
Flowing oral or intravenous administration of drugs,the concentration of the original medicine and the precursor in plasma sample were determined separately by the HPLC method which had been established.
Data analysis
Under the standard curve established by each analysis group,drug concentration was calculated in plasma.U-sing DAS2.1.1 pharmacokinetic calculation software,the main pharmacokinetic parameters of drugs were calculated through the non-compartment model.The experimental data were statistically analyzed using t test.If P<0.05,considered significant difference,P<0.01 considered highly significant difference and P>0.05 no significant difference.
Results
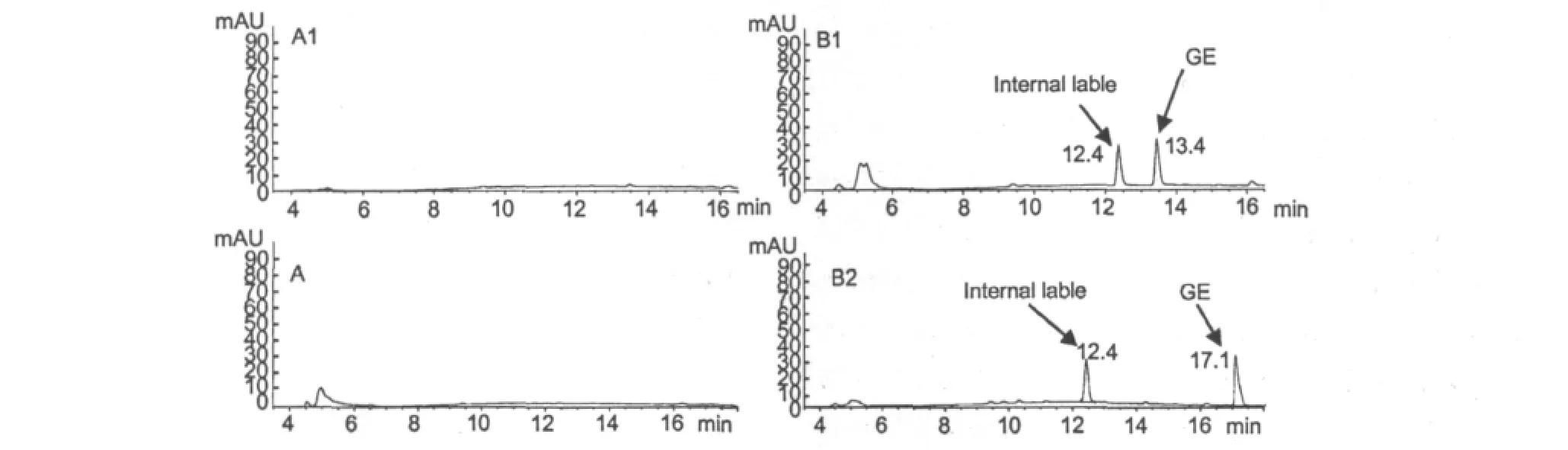
Fig.2 Representative chromatograms of GE and its derivative in plasma samples determined by HPLC method.
Analysis confirmed
Method selectivity
The rat blank plasma 50 μL were treated according to“1.3 Blood sample pretreatment”method(without internal standard).10 μL was injected into HPLC system and chromatogram Fig.2(A)was obtained.The standard solution of the lower limit of quantification of GE,GB was added into a blank plasma respectively and operated according to the same method(with internal standard),so the chromatogram Fig.2(B)were obtained,in which the retention time of GE,GB,and DZ were 13.4,17.1,12.4 min respectively.The results showed that endogenous substances in plasma do not interfere with the determination of GE,GB and DZ,also the internal standard DZ and the tested samples do not interfere with each other.The analytic period of each sample was 26.5 min.
Preparation of standard curve
Adding 10 μL of GE standard series solution to 40 μL of rat blank plasma and eddying 3 min,so their concentration(μg/mL)were 0.02,0.08,0.25,1.0,2.5,10,40 respectively.The other operation accorded to" 1.3 Blood sample pretreatment"method.To the concentration of analytes as abscissa,the ratio of peak area of analytes and internal standard as the vertical axis,carried on the return operation with the weighting least squares method,the standard curve linear regression equation obtained was Y=2×10-5X+0.0084,R2= 0.9997.The linear range of the determination of concentration of GE in plasma was 0.020~40 μg/mL according to the standard curve.The preparation of standard curve of the other drug accorded with GE in plasma.The straight linear regression equation of standard curve of GB in plasma:Y=4×10-5X+0.0231,R2= 0.9994,the linear range is 0.025~25.6 μg/mL.
Adding 10 μL of standard solution of GE to 40 μL of blank plasma,the sample of the equivalent with GE 0.02 μg/mL was obtained.Carried on 5-sample analysis to the sample,based on the standard curve of the same day,the concentration of each sample was obtained.The determination of the lowest limit of quantification of GB accorded with GE.The results show that the lowest limit of quantification of GE and GB in plasma were 0.02 μg/mL and 0.0125 μg/mL with HPLC method respectively.The with-day precision(relative standard deviation,RSD)of GE and GB in lowest limit of quantification concentration was 13.7%and 14.3% respectively,and the accuracy(RE)in this concentration was 9.2%and-8.0%respectively.
The precision and accuracy of methods
Taking 40 μL blank plasma,the low,medium and high concentration of three quality control(QC)samples of GE were prepared according to"2.1.2 Preparation of standard Curve"method.Each concentration was completed for 6-sample analysis and determined for three days continuously,and their standard curve was prepared at the same time.Calculating the concentration of QC samples and comparing with the added concentration,the accuracy and precision of determination method of each component was sought.The determination method of RE and RSD of GB accorded with the determination of GE.Experimental data indicate that the with-day and between-day precision of GE,GB were less than 12%,and accuracy were at±10%.The determination method of compounds tested in plasma is in compliance with the relevant international standards requirements[8].
The extraction recovery of sample
According to"2.1.2 Prepation of standard Curve" method,the QC samples of GE,GB and DZ were prepared and the extraction recovery of these samples were reviewed.The results showed that the extraction recovery of three kinds of concentration of components were over 70%in general,only the recovery of GE was 66.1%at high concentration.
The stability of sample
According to“2.1.2 Preparation of standard Curve”method,the low,medium and high concentration of three QC samples of GE,GB were prepared to study the stability of samples in three different preservation conditions.The results showed that the tested compounds were stable at the-70℃ during 30 d,as well as after three freezing-defrosting circulation.The plasma sample solutions extracted were stable after 24 h at room temperature.The RE of various concentration groups were-10.2% ~12.8%.
The drug metabolism research in vivo
After the Wistar rat was being oral administration of 100 mg/kg GB,its plasma samples were analysised using HPLC.The experimental results as shown in Fig.3.The results showed that GE can be detected in rat plasma obviously after oral administration of GB to Wistar rats.Thus GB has the natures of prodrug.
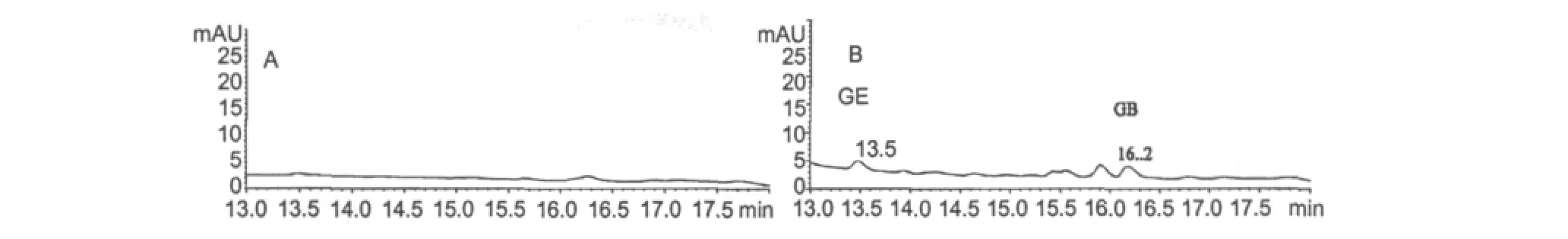
Fig.3 HPLC of compounds in rat plasma after oral administration of 100 mg/kg GB to Wistar rats.(A-blank plasma.)

Fig.4 The mean plasma concentration-time curve of GE following oral administration of 40 mg/kg GE to Wistar rats(n=5).Each value represents the mean±SD of five individual values.
Pharmacokinetics
Pharmacokinetics of the parent drug
Pharmacokinetics of parent drug GE in rats in vivo have been studied in detail in the literature[9].To accurately investigate its relative bioavailability in prodrug,we have also studied its pharmacokinetics after its intragastric administration to rats under the same experimental conditions.
Following oral administration with 40 mg/kg GE to rats,its mean plasma concentration-time curve see Fig.4.The blood drug concentration data were fitted with compartment model and pharmacokinetic parameters were calculated by the DAS 2.1.1.It found the dynamic processes of GE consistent with a compartment model after intragastric administration of GE to rats.The pharmacokinetic parameters respectively were:t1/2,8.18±5.30 h;Cmax,3511±2408 ng/mL;Tmax,1.20± 1.60 h;AUC0-t,15550±1649 ng.h/mL;AUC0-∞,18665±2879 ng.h/mL.By the drug concentrationtime curves and pharmacokinetic parameters,it can be found that the cure of GE appears double peaks after intragastric administration.The tmax1,tmax2of these two peaks respectively were 10 min and 4 h,and the Cmax1and Cmax2respectively were 3029 and 1494 ng/mL.Half-life was 8.18 h or so,which showed that GE can be maintained a longer time in the blood and maintain its efficacy.
Pharmacokinetics of prodrug
The pharmacokinetic behaviors were studied after intragastric or intravenous administration of GB to rats with established HPLC method.After oral and intravenous administration with 40 mg/kg dose of GB to rats,the mean plasma concentration-time curves of GE and GB see Fig.5 and 6 respectively.Blood drug concentration data were fitted with compartment model by the DAS 2.1.1 software and pharmacokinetic parameters were calculated.It found that the dynamic processes of GE consistent with a compartment model after intragastric administration of GB to rats and that the dynamic processes of GE were consistent with a compartment model after intravenous administration of GB to rats.Pharmacokinetic parameters see Table 1.

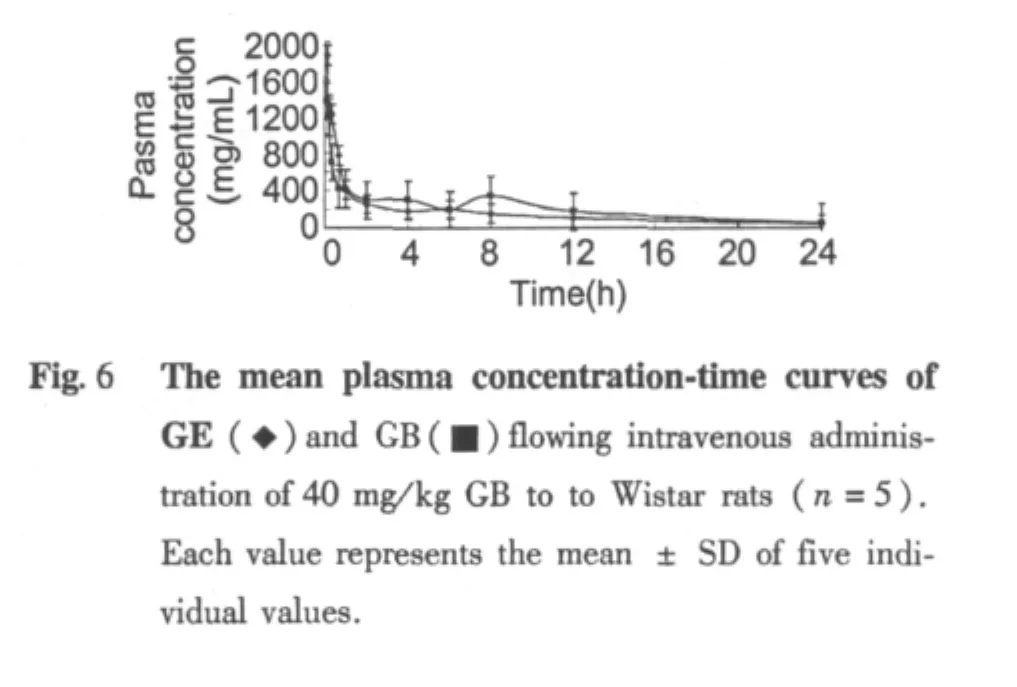
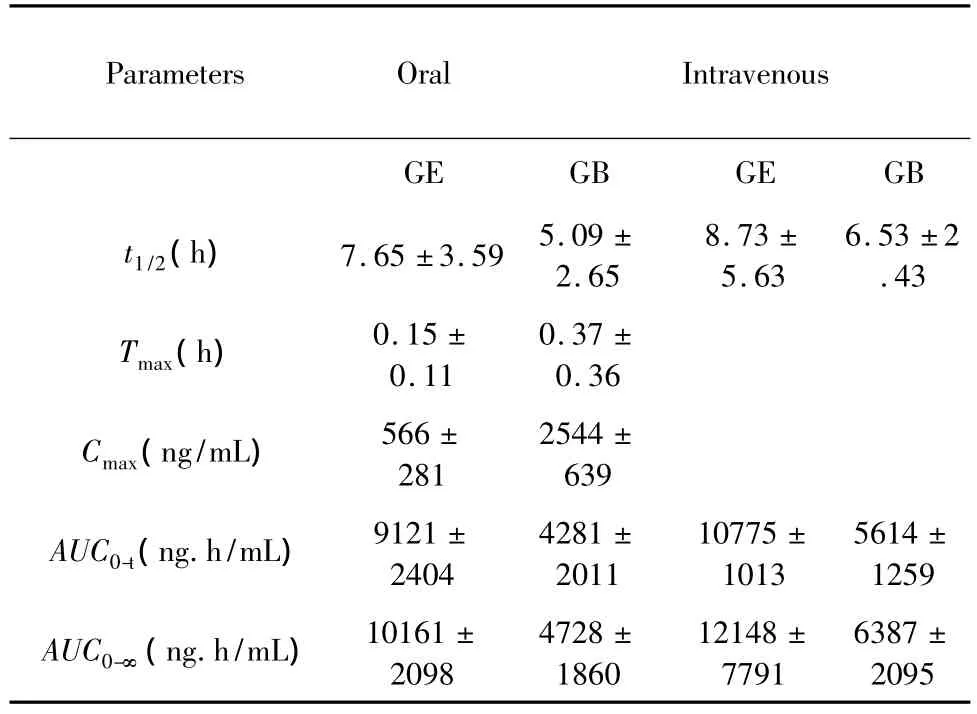
Table 1 The mean non-compartmental pharmacokinetic parameters of DZ and GB in Wistar rats after single oral and intravenous administration of 40 mg/kg GB(n=5).Each valuerepresents the mean±SD for five individual values.
By the drug concentration-time curves of Fig.5 and 6 and Table 1,it can be found that the Tmaxof GE is about 0.15 h after orally administration of GB to rats.It indicated that GB was rapidly absorbed in vivo and hydrolyzed by enzymes to the parent drug GE.By the drug concentration-time curves,it can be found that the cure of GB appears double peaks after intragastric administration of GB.The Tmax1,Tmax2 of these two peaks respectively were 5 min and 8 h.The Cmax1and Cmax2respectively were 1359 and 351 ng/mL.It may be related to that GB has large molecular weight or enterohepatic cycle.Comparison the plasma concentration-time curve area AUC0-∞。of intravenous and intragastric administration of GB,a small difference between the two showed that first-pass effect would be very small when GB was being orally administration.
Pharmacokinetic characteristics of prodrug
Fllowing intragastric administration of prodrug,the Tmax,t1/2and AUC0-∞of GE that come from hydrolysis of prodrug GB see Table 1.The dose of prodrug converted into the equivalent of the same dose(Dtest)of GE by equimolar respectively.The relative bioavailability (Fr)of GE in prodrug was calculated by the formula (1),see Table 2.

*:Dstandard:the oral dose of the parent drug,Dtest:the dose of the parent drug converted by oral prodrug.
AUCstandard:the area under the concentration-time curve of parent drug flowing oral administration of parent drug,AUCtest:the area under the concentration-time curve of parent drug flowing oral administration of prodrug.

Table 2 The relative bioavailability of GE after single oral administration of GB to Wistar rats.
After intragastric administration of prodrug,the Tmaxof GE from the prodrug that was in one hour,is shorter than Tmaxof parent GE.Perhaps due to the large molecular weight,after intragastric administration of the prodrug GB,it occured hepatobiliary circulation.
According to the relative bioavailability,drugs in rats release the amount of GE in the order for the GB>GE.It indicated that the kinetic behavior of GB compared with GE is known as a great improvement by proper structural modification.
After intragastric administration of the prodrug,the Tmaxof GE that come from the hydrolysis of the prodrug was shorter than Tmax(1.2 h)of parent GE.After intragastric administration of GB,the half-live was 7.65 h.From Fig.1,compared to the GE,4’and 7-OH of GB all were substituted by benzene sulfonate group.Its lipophilic is enhanced,and it may be better to avoid the first pass effect.Its relative bioavailability is larger than GE in expermients.GB maybe have further research value.
Discussion
The aim of design and synthesis of prodrugs was to solve the specific problems in pharmacy or pharmacology and give the useful nature to drugs.The main objective of sulfonic acid ester-modified of GE was to improve its absorption in vivo and block first-pass effect,thus to enhance its bioavailability and pharmacological effects possibly.Polyhydroxy flavonoids,mainly referring to natural active product GE,DZ,etc.,have different degree of pharmacokinetic shortcomings,mainly the first-pass effects,intestinal flora decomposition,fast elimination in vivo,etc.,and the same time also poor water solubility in general[10,11].The deficiencies in pharmacokinetics and poor water solubility are main reasons for their low bioavailability.So prodrug modifition of such polyhydroxy flavonoids must start from improving these two areas weaknesses.
According to affecting factors of the drug bioavailability,combined with the physical and chemical properties of polyhydroxy flavonoids,as well as their basic transmission characteristic in vivo,the prodrug design for multi-hydroxyflavone was carried out mainly in lipophilic aspect.The big sulphonate group introduced in molecular could seal up its easily metabolized phenol hydroxyl,improve its solubility and amphipathicity,enable to have the good biomembrane endophilicity,increase the absorption rate,avoid the metabolism before circulation,and enhance the bioavailability.
This paper had carried on prodrug screening to the new sulphonate derivative of GE,and conducted the medicine dynamics research of the prodrug confirmed.The experiments indicated that the bioavailability of GE flowing oral administration of GB were better than that of GE following oral administration of GE.This explained that thinking of optimizing kinetic property of GE by sulphonate modification to enhance the oral bioavailability is successful.The novel sulfonic acid ester derivative was prodrug of GE that was confirmed by metabolism screening studies in rats.Its pharmacokinetic studies had showed that the prodrug GB had good oral bioavailability.The idea that we adopted the structural modification of the hydroxyl group of GE to optimize the pharmacokinetic properties,block first-pass metabolism and thus improve the oral bioavailability is feasible.
1 Coward L,Barnes NC,Setchell KDR,et al.Genistein,daidzein,and their β-glcoside conjugates:antitumor isoflavones in soybean foods from American and asian diets.J Agric Food Chem,1993,41:1961-1967.
2 Aedin C,Bryn H,Rosa M.Isoflavones,lignans and stilbenesorigins,metabolism and potentialimportancetohuman health.J Sci Food Agric,2000,80:1044-1047.
3 Holder CL,Churchwell MI,Doerge DR.Quantification of soy isoflavones,genistein and daidzein,and conjugates in rat blood using LC/ES-MS.J Agric Food Chem,1999,47:3764-3770.
4 Huang J,Luo Q,Li XL,et al.Hyperglycemia Lowering Effect of Soybean Isoflavone(SI)on Alloxan-induced Diabetic Mice.Food Sci,2004,25:166-170.
5 Barne S,Sfakinos J,Coward L,et al.Soy isoflavonoids and cancer prevention.Underlying biochemical and pharmacological issues.Adv Exp Med Biol,1996,401:87-100.
6 Andlauer W,Kolb J,Stehle P,et al.Absorption and metabolism of genistein in isolated rat small intestine.J Nutr,2000,130:834-846.
7 Peng Y,Deng ZY,Lang SJ,et al.Preparation and crystal structure of genistein benzensulfonate prodrugs.J Chem Res,2008,10:555-558.
8 Shah VP,Midha KK,F(xiàn)indlay JW,et al.Bioanalytical method validationa revisit with a decade of progress.Pharm Res,2000,17:1551-1557.
9 Setchell KD,F(xiàn)aughnan MS,Avades T,et al.Comparing the pharmacokinetics of daidzein and genistein with the use of13C-labeled tracers in premenopausal women.Am J Clin Nutr,2003,77:411-419.
10 Hur HG,Lay JO,Jr Beger RD,et al.Isolation of human intestinal bacteria metabolizing the natural isoflavone glycosides daidzin and genistin.Arch Microbiol,2000,174:422-428.
11 Hollman PC,Katan MB.Bioavailability and health effects of dietary flavonols in man.Arch Toxicol Suppl,1998,20:237-248.
