海金子中檉柳素-3-O-蕓香糖苷和蘆丁的分離及含量測定
2012-11-26 07:42:12彭一波劉玉琴王實強
湖南中醫藥大學學報 2012年7期
彭一波,劉 陽,李 娜,劉玉琴,謝 誼,王實強
(1.湖南中醫藥大學,湖南 長沙410208;2.湖南省中醫藥研究院中藥研究所,湖南 長沙410013)
海桐花科(Pittosporaceae) 海桐花屬(Pittosporum Banks)植物海金子(Pittosporum.illicioides Mak.)分布于福建、浙江、江蘇、安徽、江西、湖北、湖南、貴州等省,國外產地有日本[1]。海金子的根民間用于風濕性關節炎、坐骨神經痛、骨折、胃痛、牙痛、高血壓及神經衰弱,還具有良好的殺精子作用[2-3]。海金子葉可用于蛇咬傷,瘡癤腫毒,過敏性皮炎和外傷出血[3]。海金子的干燥種子稱為“山枝仁”,被貴州和四川兩省的地方標準收載,具有清熱利濕、生津止咳、收斂止瀉的功效。用于心煩,口渴咽痛,痢疾,腸炎,白帶,體倦乏力等[4-5]。海金子在四川幾乎都為野生,多生長在懸崖峭壁或斜坡上,采集十分困難,加上利潤微薄,少有人愿意采集,導致商品來源不足。
為了更深入地研究海金子的活性物質,從海金子葉中提取、分離得到活性化合物檉柳素-3-O-蕓香糖苷和蘆丁,這兩種化合物在海金子中首次發現。目前關于海金子中這兩種化合物的高效液相色譜分析方法未見文獻報道。本文建立了快速預處理、分離效果好的高效液相色譜分析法,測定不同采集時間、海金子不同部位兩種化合物的含量。為開發和利用海金子提供參考依據。
1 實驗材料
1.1 實驗儀器
島津LC-10ATvp 高效液相色譜儀(日本島津公司),N2000 雙通道色譜工作站 (浙江大學智能信息工程研究所);島津UV-2450 型紫外分光光度儀(日本島津公司);INOVA-400 核磁共振儀 (Varian 公司);Avatar 370 FT-IR 型紅外光譜儀(Thermo Nicolet 公司);LCQ Advantage 型質 譜 儀(Thermo Finnigan 公司);C10558 型TLC visualizer 成像儀(瑞士卡瑪公司);SZ-93A 雙蒸餾水器(上海亞榮生化儀器廠);KQ2200B 型超聲波清洗器(昆山市超聲儀器有限公司);AE240 型分析天平 (METTLER 公司)。
1.2 藥品與試劑
檉柳素-3-O-蕓香糖苷對照品(自制),HPLC 測定含量大于98%;蘆丁對照品(中國藥品生物制品檢定所提供,含量測定用,批號100080-200707);甲醇、磷酸為色譜純;其他試劑均為分析純;水為雙重蒸餾水。海金子樣品由課題組成員從保靖、吉首、花垣采集,經湖南省中醫藥研究院中藥研究所生藥室鑒定,為海金子(Pittosporum.illicioides Mak.)全草,取其葉、莖皮、果實陰干,供樣品含量測試用。
2 方法與結果
2.1 提取分離
將海金子(花垣8月)葉(500 g)的70%乙醇提取物,依次用石油醚、水飽和正丁醇萃取,回收正丁醇得提取物(3.5 g)。采用硅膠柱進行分離,從石油醚∶乙酸乙酯(1∶1,V/V)洗脫液中得到深黃色顆粒狀晶體,重結晶得化合物Ⅰ(約200 mg),從乙酸乙酯∶甲醇=(4∶1,V/V) 洗脫液中得到黃色粉末狀結晶,重結晶得化合物Ⅱ(約300 mg)。
2.2 結構鑒定
化合物Ⅰ進行UV、1H-NMR、13C-NMR、ESIMS、IR 光譜鑒定,化合物Ⅱ進行UV、TLC、HPLC 鑒定。
2.2.1 化合物Ⅰ的波譜數據及鑒定
UV λMeOmaxHnm(logε):254(4.32),356(4.26),可初步判斷有黃酮化合物基本母核。
ESI -MS m/z:623 [M -H]-。IRνKBrmax/cm:3382,1652,1604,1500,1452,1360,1291。
1H NMR(400 MHz,DMSO-d6)δ:6.44(d,1H,J=1.6 Hz,H-6),6.21 (d,1H,J=1.6 Hz,H-8),7.86(d,1H,J=1.2 Hz,H-2′),7.52(dd,1H,J=8.4 Hz,1.2Hz,H-6′),6.91(d,1H,J=8.4 Hz,H-5′),3.84(s,3H,4′-OCH3),5.44 (d,1H,J=7.2 Hz,glc-H-1"),3.22~3.41(m,6H,glc-H-2"~6"),0.98 (d,3H,J=6.0 Hz,rha-CH3)。
13C NMR(100 MHz,DMSO-d6)δ:156.8(C-2),133.3(C-3),177.7(C-4),161.5(C-5),99.0(C-6),164.4(C-7),94.1(C-8),156.8(C-9),104.3(C-10),121.4(C-1′),113.6(C-2′),147.2(C-3′),149.7(C-4′),115.6(C-5′),122.6(C-6′),101.5(C-1"),76.7(C-2"),76.2(C-3"),70.6(C-4"),74.6(C-5"),67.1(C-6"),101.2(C-1'''),70.4(C-2'''),70.9(C-3'''),72.1(C-4'''),68.6(C-5'''),18.0(C-6''')。以上波譜數據與檉柳素-3-O-蕓香糖苷的文獻數據[6]一致,故鑒定為檉柳素-3-O-蕓香糖苷。
2.2.2 化合物Ⅱ的結構鑒定
薄層鑒別:取化合物Ⅱ10.1 mg,用甲醇定容至10 mL,制成化合物Ⅱ溶液;取蘆丁對照品10.5 mg,用甲醇溶解定容至10 mL,即得蘆丁對照品溶液。照薄層色譜法[7]試驗,吸取上述兩種溶液各5 μL,分別點于同一硅膠G 薄層板上,分別以不同的展開劑展開:(1)二氯甲烷∶甲醇∶甲酸(15∶7∶0.5);(2)三氯甲烷∶甲醇∶水(2∶1∶0.3)下層20 mL 加甲酸0.2 mL;(3)三氯甲烷∶丙酮∶冰醋酸(2∶1.5∶0.2)。展距為15 cm,取出,晾干。置紫外光燈(365 nm)下檢視。結果化合物Ⅱ溶液的色譜中,在與蘆丁對照品相應的位置處顯黑色斑點。
紫外鑒別:向化合物Ⅱ的甲醇溶液(26.8 μg/mL)分別加入甲醇鈉、乙酸鈉、乙酸鈉/硼酸、三氯化鋁及三氯化鋁/鹽酸診斷試劑,試驗數據(λmaxnm):256,266 sh,296 sh,358 (MeOH);272,328,410 (NaOMe);272,302 sh,420 (AlCl3);268,300,366 sh,398(AlCl3/HCl);272,324,382 (NaOAc);262,298,378(NaOAc/H3BO3)。將上述各種UV 圖譜與文獻[8]比較,結果與蘆丁一致。
高效液相色譜鑒別:取蘆丁對照品適量,制成100 μg/mL 對照品溶液;取化合物Ⅱ適量,制成100 μg/mL 的化合物Ⅱ試液,在“2.3.1”色譜條件下分別檢測。發現化合物Ⅱ與蘆丁對照品在相同的保留時間(15.4 min)出峰。綜上,化合物Ⅱ鑒定為蘆丁。
2.3 含量測定
2.3.1 色譜條件與系統適應性 采用Agela Technologies Promosil C18(200 mm×4.6 mm,5 μm)色譜柱;流動相為甲醇-0.4%磷酸水溶液(34∶66);流速為1.0 mL/min;檢 測 波 長 為254 nm;柱 溫 為40 ℃;理論塔板數按檉柳素-3-O-蕓香糖苷計不低于5 000。結果見圖1。
2.3.2 對照品溶液的制備 稱取檉柳素-3-O-蕓香糖苷對照品和蘆丁對照品適量,精密稱定,用50%甲醇溶解并定量稀釋成含檉柳素-3-O-蕓香糖苷100 μg/mL,蘆丁100 μg/mL 的混合對照品溶液。
2.3.3 供試品溶液的制備 藥材粉碎(過一號篩),取粉末0.5 g,精密稱定,置具塞錐形瓶中,精密加入50%甲醇25 mL,稱定重量,超聲提取30 min,放冷至室溫,用50%甲醇補足失去的質量,搖勻,用微孔濾膜(0.45 μm)濾過,取續濾液即得。

圖1 對照品(A)及海金子藥材葉(花垣8月)供試品(B)HPLC 圖
2.3.4 線性關系的考察 精密稱取檉柳素-3-O-蕓香糖苷對照品和蘆丁對照品適量,用50%甲醇分別配成每1 mL 含檉柳素-3-O-蕓香糖苷0.254 4 mg和每1 mL 含蘆丁0.340 8 mg 的2 份母液,分別吸取檉柳素-3-O-蕓香糖苷母液2、3、5、7、9 mL于10 mL 容量瓶;分別吸取蘆丁母液2、3、5、7、9 mL 于10 mL 容量瓶,用50%甲醇稀釋至刻度。分別吸取以上10 個稀釋液和2 個母液各5 μL 注入液相色譜儀。以對照品進樣量(μg)為橫坐標,色譜峰面積積分值(mv)為縱坐標,繪制標準曲線。結果見表1。

表1 檉柳素-3-O-蕓香糖苷和蘆丁的線性關系考察
2.3.5 精密度實驗 精密吸取海金子(花垣8月)葉供試品溶液,按上述色譜條件和測定方法,連續進樣6 次,以檉柳素-3-O-蕓香糖苷和蘆丁峰面積積分值計算,RSD 分別為1.71%和1.82%,結果表明本法精密度良好。
2.3.6 穩定性實驗 精密吸取同一份海金子(花垣8月)葉供試品溶液,按上述色譜條件和測定方法,分別在0、2、4、6、8、10、12 h 進樣,測定色譜峰面積,以檉柳素-3-O-蕓香糖苷和蘆丁峰面積積分值計算,RSD 分別為1.52%和1.73%,結果表明供試品在12 h 內基本穩定。
2.3.7 重復性實驗 按“2.3.3”方法制備海金子(花垣8月)葉樣品5 份,在“2.3.1”色譜條件下分析,測得檉柳素-3-O-蕓香糖苷的平均含量為0.54%,RSD為1.76%,蘆 丁 的 平均 含 量 為0.47%,RSD 為1.34%,結果表明該方法重現性良好。
2.3.8 加樣回收率試驗 精密稱取已知檉柳素-3-O-蕓香糖苷和蘆丁含量的海金子(花垣8月)葉粉末(過一號篩)12 份,每份0.25 g,分兩組,一組精密加入檉柳素-3-O-蕓香糖苷,另一組精密加入蘆丁對照品適量,按“2.3.3”方法制備待測液,各精密吸取5 μL 進樣,在“2.3.1”色譜條件下分析測定,計算回收率。測定結果見表2~3。
2.3.9 樣品含量測定 取不同采集時間的海金子葉、莖皮、果實的粉末(過一號篩),按“2.3.3”項下方法制備供試品溶液,精密吸取對照品溶液和供試品溶液各5 μL,注入液相色譜儀,依照“2.3.1”色譜條件測定,采用外標法計算含量,結果見表4。
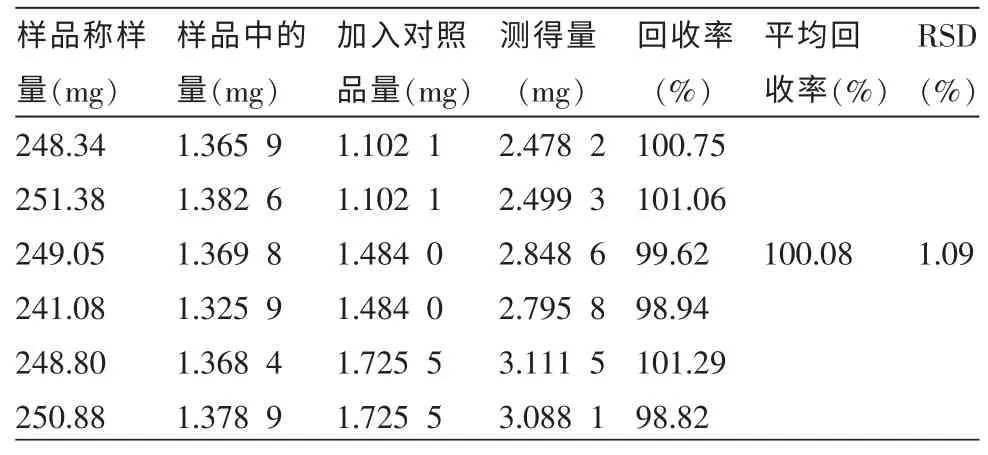
表2 檉柳素-3-O-蕓香糖苷回收率試驗 (n=6)
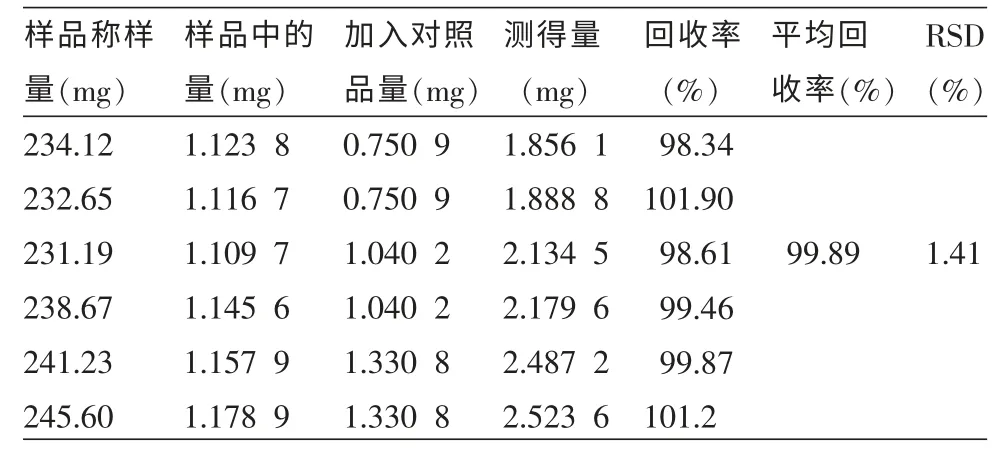
表3 蘆丁回收率試驗 (n=6)

表4 不同采集地、時間海金子葉、莖皮、果實中檉柳素-3-O-蕓香糖苷和蘆丁含量 (n=2,%)
3 討論
3.1 色譜條件選擇
色譜柱:本實驗分別使用了伊利特Hypersil BDS C18(250 mm×4.6 mm,5 μm) 柱,Agela Technologies Promosil C18(200 mm×4.6 mm,5 μm) 柱,Waters Symmetry C18(3.9 mm×150 mm,5 μm)柱,發現 Agela Technologies Promosil C18(200 mm ×4.6 mm,5 μm)柱,分離情況較好。
流動相的選擇:甲醇-0.2%磷酸[9],拖尾,適當增加磷酸百分含量,甲醇-0.4%磷酸,峰形好。摸索流動相比例,甲醇-0.4%磷酸水溶液(34∶66),分離情況較好。
檢測波長的選擇:檉柳素-3-O-蕓香糖苷的紫外光譜圖有兩個特征吸收峰,最大吸收波長分別為254 nm 和356 nm。因254 nm 處 的ε 值 大 于356 nm 處的ε 值,故選用254 nm 為檢測波長。
柱溫的選擇:比較柱溫30、35、40 ℃時的色譜圖,在40 ℃時分離度符合要求、理論塔板數較高。
3.2 樣品處理方法的選擇
提取溶劑考察:取海金子(花垣8月)葉粉末(過一號篩),8 份,每份0.5 g,分別用不同的溶劑提取:95%、70%、50%、20%乙醇及100%、70%、50%、20%甲醇,其他提取和測定條件相同。測得樣品含量,結果50%甲醇提取最完全。
提取方法考察:取海金子(花垣8月)葉粉末(過一號篩),2 份,每份0.5 g,分別回流、超聲30 min,其他提取和測定條件相同。測得樣品含量,結果超聲效果比回流好。
提取時間考察:取海金子(花垣8月)葉粉末(過一號篩),4 份,每份0.5 g,分別超聲20、30、45、60 min 放冷,其他提取和測定條件相同。測得樣品含量,結果30 min 樣品可提取完全。
3.3 結果分析
蘆丁結構鑒定中,由于有中檢所對照品提供,采用液相色譜結合薄層色譜進行鑒別,比照樣品與對照品的保留時間與Rf 值確定該化合物為蘆丁;而檉柳素-3-O 蕓香糖苷無法定對照品,故選用波譜技術進行結構鑒定。
從表4中可以看出海金子葉、莖皮、果實中檉柳素-3-O-蕓香糖苷和蘆丁含量差異顯著,兩個化合物葉含量最高;不同采集時間海金子檉柳素-3-O-蕓香糖苷和蘆丁含量差異顯著,其中檉柳素-3-O-蕓香糖苷8月含量較低,蘆丁3月含量較低。本文采用HPLC 測定幾個不同采集時間海金子葉、莖皮、果實中檉柳素-3-O-蕓香糖苷和蘆丁的含量,為海金子的開發利用提供了實驗依據。
[1]中國科學院《中國植物志》編輯委員會.中國植物志[M].北京:科學出版社,2005,53(2):16.
[2]秦松云,吳 紅,鐘明芳.四川省海桐花屬藥用植物的種類與分布[J].資源開發與保護,1991,8(4):265-267.
[3]聶田田,白 虹,王元書.海桐花屬植物化學成分及藥理活性研究進展[J].齊魯藥事,2010,29(6):354-357.
[4]羅達尚.新修晶珠本草[M].成都:四川科學技術出版社,2004:513.
[5]張 俊,蔣桂華,敬小莉.山枝仁質量標準初步研究[J].中藥與臨床,2011,2(1):15-19.
[6]El-Sayed N H,Abu Dooh A M,El-KhrisySAM,et al.Flavonoids of Cassia italica[J].Phytochemistry,1992,31(6):2 187-2 189.
[7]中華人民共和國衛生部藥典委員會.中華人民共和國藥典[S].北京:中國醫藥科技出版社,2010:附錄34.
[8]匡海學.中藥化學[M].北京:中國中醫藥出版社,2003:157.
[9]王錦軍,楊成雄.馬齒莧中黃酮化合物的高效液相色譜分析[J].解放軍藥學學報,2010,26(4):345-347.
