氧分子在黃鐵礦和方鉛礦表面的吸附
2012-11-24 12:53:12李玉瓊陳建華藍麗紅
中國有色金屬學報 2012年4期
李玉瓊,陳建華,藍麗紅,,郭 進
(1. 廣西大學 化學化工學院,南寧 530004;2. 廣西大學 資源與冶金學院,南寧 530004;3. 廣西民族大學 化學與生態工程學院,南寧 530006;4. 廣西大學 物理科學與工程技術學院,南寧 530004)
氧分子在黃鐵礦和方鉛礦表面的吸附
李玉瓊1,陳建華2,藍麗紅1,3,郭 進4
(1. 廣西大學 化學化工學院,南寧 530004;2. 廣西大學 資源與冶金學院,南寧 530004;3. 廣西民族大學 化學與生態工程學院,南寧 530006;4. 廣西大學 物理科學與工程技術學院,南寧 530004)
采用密度泛函理論對氧分子在黃鐵礦和方鉛礦表面的吸附進行研究。計算結果表明:黃鐵礦和方鉛礦表面經歷了較小的弛豫;氧分子在黃鐵礦和方鉛礦表面都呈解離吸附狀態,且在黃鐵礦表面的吸附能遠低于在方鉛礦表面的吸附能;在黃鐵礦表面上,氧原子分別與鐵原子和硫原子鍵合,電子由鐵原子和硫原子轉移到氧原子上,主要由硫的3p態、氧的2p態和鐵的3d態參與反應,鐵與氧之間形成d→p反饋鍵,而在方鉛礦表面上,氧原子只與硫原子鍵合,主要由硫的3p態、氧的2p態和鉛的6p態參與反應,未形成反饋鍵;氧吸附后黃鐵礦表面產生鍵合的鐵原子和氧原子都產生自旋現象,而方鉛礦表面原子及吸附的氧原子仍然是低自旋態的。
黃鐵礦;方鉛礦;氧分子吸附;密度泛函理論
有色金屬硫化礦浮選是一個電化學過程,礦物表面的氧化對其浮選行為具有決定性的影響,硫化礦物的浮選行為與表面氧化之間存在密切的關系[1-3]。已有研究證實:捕收劑黃藥在硫化礦物表面的吸附是一個電化學反應過程,黃藥在硫化礦表面發生陽極氧化生成金屬黃原酸鹽或雙黃藥,氧分子在礦物表面發生陰極還原。氧分子在硫化礦表面吸附對黃藥捕收的影響非常大。另外,硫化礦無捕收劑浮選主要是通過氧化來控制礦物表面產物形成疏水元素硫和親水硫酸鹽,實現礦物的浮選和抑制[4-5]。因而氧分子在硫化礦浮選中的作用成為硫化礦浮選研究中最重要的理論問題之一。
黃鐵礦和方鉛礦是浮選電化學過程中最具代表性的兩種硫化礦,捕收劑黃藥分別在黃鐵礦表面和方鉛礦表面形成疏水的雙黃藥和金屬黃原酸鹽,代表硫化礦捕收作用的兩種典型電化學機制,另一方面,方鉛礦具有良好的無捕收劑浮選行為,而黃鐵礦則較差。不論是黃藥捕收劑浮選還是無捕收劑浮選,都與氧分子在這兩種礦物表面的還原密不可分。目前的研究多集中在硫化礦表面氧化動力學和表面化學方面[6-15]。RAIKAR等[16]采用自然黃鐵礦作為研究對象,發現將黃鐵礦暴露在氧氣中鐵組分將出現嚴重的氧化現象,而ROSSO等[17]也發現真空解理的黃鐵礦與氧作用后表面形成了Fe—O鍵,此外,KNDELEWICZ等[18]則發現,黃鐵礦僅暴露給水沒有導致硫組分氧化,而單獨暴露給氧則導致了硫組分的氧化。而對于氧分子在黃鐵礦和方鉛礦表面上的吸附方式、吸附構型以及礦物表面電子轉移情況等詳細的吸附機理卻缺少研究。王淀佐等[19]采用分子軌道法針對黃鐵礦和方鉛礦的氧化進行了研究,但他們只采用單層原子來模擬礦物表面,因此,其計算結果很難與實際表面吸附相結果一致。ROSSO等[17]采用試驗和叢簇計算相結合的方法,表明氧在黃鐵礦表面解離吸附。SUN等[20]采用密度泛函方法簡單研究了氧分子在黃鐵礦表面的吸附,但沒有考慮黃鐵礦和氧分子的自旋。
本文作者通過構造含多層原子的礦物表面層晶模型,采用密度泛函理論對黃鐵礦和方鉛礦表面弛豫和氧分子在表面上的吸附方式、吸附能、表面電荷分布和電子轉移及表面態進行詳細的研究,研究結果對進一步闡明氧氣在硫化礦電化學浮選中的作用具有重要意義。
1 計算方法和模型
1.1 計算方法
本文作者采用基于密度泛函理論和平面波贗勢方法的CASTEP軟件[21-22]進行計算。交換關聯泛函采用廣義梯度近似(GGA)下的PW91梯度修正近似。平面波截斷能經過測試后對黃鐵礦選取270 eV,對方鉛礦選取280 eV。表面由優化過的體相切出,并在吸附前進行優化,為了消除相鄰吸附分子之間的相互影響,分別對黃鐵礦和方鉛礦采用(2×2)和(4×2)表面層晶模型。進行氧分子吸附計算時,將氧分子置于不同的位置上,如垂直置于硫位、鉛位、鐵位和穴位以及平躺等方式,通過優化構型、計算吸附能,確定最穩定的吸附模式。對價電子和離子實的相互作用勢的描述采用超軟贗勢(USP)[23],Brillouin區的積分計算分別對黃鐵礦采用 2×2×1和對方鉛礦采用 1×2×1的Monkhorst-Pack(MP)k點網絡[24-25]。計算的價電子構型為Fe 3d64S2,S 3s23p4和Pb 6s26p2。幾何優化采用BFGS算法,對黃鐵礦進行優化的收斂標準如下:能量收斂標準為 2.0×10-5eV·atom-1,原子位移的收斂標準為0.000 2 nm,原子間作用力的收斂標準為0.8 eV·nm-1,晶體內應力收斂標準為0.1 GPa;自洽迭代收斂精度為 2.0×10-6eV·atom-1;除對方鉛礦進行優化的收斂標準除原子間作用力的收斂標準采用 0.5 eV·nm-1外,其余收斂標準與黃鐵礦相同。對含不同原子層及真空層厚度的表面進行了計算,以獲得較穩定的表面,根據最終的測試結果確定:對于黃鐵礦,采用15層原子層以及15 ?埃真空層厚度的表面模型(考慮到需要吸附氧分子,所以真空層取值大一點),并在吸附過程中固定基底9層原子層,對表面6層原子進行弛豫;對于方鉛礦,采用8層原子層,真空層厚度的采取與黃鐵礦一致,吸附過程中固定基底5層原子層,對表面3層原子進行弛豫;對于氧分子,則將其放入一個1.5 nm×1.5 nm×1.5 nm的盒子中,計算時采用Gamma點。所有計算都采用自旋極化方法。
1.2 計算模型
1.3 吸附能計算
氧分子在黃鐵礦和方鉛礦表面的吸附能按下式定義:
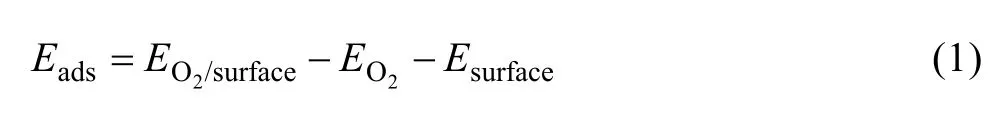
其中:Eads為氧分子吸附后的吸附能; EO2/surface為氧分子在表面吸附后體系的總能; EO2和 Esurface分別為吸附前氧分子和表面的總能。吸附能越低,吸附越穩定,反之則穩定性降低。
2 結果與討論
2.1 黃鐵礦和方鉛礦表面結構弛豫
計算得到黃鐵礦和方鉛礦的晶格常數分別為0.542 1和0.601 8 nm,分別與實驗值0.541 7和0.593 6 nm非常接近[26-27],優化后的氧分子中 O—O鍵長為0.124 1 nm,也與實驗值0.120 9 nm非常接近,表明計算是可靠的。表1和表2所列分別為弛豫后黃鐵礦和方鉛礦表面幾層原子的配位數及位移,其中負號表明原子沿軸的負方向弛豫,反之則沿軸正方向弛豫。在黃鐵礦表面,表面第一層S原子向表面內部弛豫;最明顯的弛豫是第二層的表面鐵原子,向內部弛豫約0.01 nm;第三層中的S原子則向表面弛豫。原子僅在頂部3層產生了明顯的弛豫,第四層至第六層原子經歷了微小的位移,第七層至第九層原子的弛豫可以忽略不計。這與 ROSSO等[28]及 CHATURVEDI等[29]的實驗測試結果一致,也與 HUNG 等[30]的計算結果一致。對于方鉛礦表面,第一層的硫原子和鉛原子向表面內部弛豫,而第二層的原子都沿z軸方向表面外部弛豫,且這一層的硫原子和鉛原子弛豫最為明顯,第三層原子都向表面內部弛豫,且弛豫較小。
表面弛豫計算表明,黃鐵礦和方鉛礦表面解理后都發生了不同程度的表面弛豫,但沒有產生明顯的表面重構作用,且僅有頂部3層原子的弛豫略微明顯,更低層原子的弛豫非常小。

圖1 (2×2)黃鐵礦(100)表面層晶模型以及(4×2)方鉛礦(100)表面層晶模型Fig. 1 Slab models of (2×2) pyrite (100) surface (a) and (4×2) galena (100) surface (b)

表1 黃鐵礦表面原子配位及位移Table 1 Atomic coordination and displacements of pyrite surface
2.2 氧分子在黃鐵礦和方鉛礦表面的吸附
為了確定氧分子在黃鐵礦和方鉛礦表面的吸附方式,分別對氧分子在各個吸附位進行了測試,結果如圖2和圖3所示。并計算了吸附能,計算結果如表3和表4所列。吸附能計算結果表明,在黃鐵礦表面,氧分子在頂部硫位(見圖 2(a))、平行于 S—S鍵(見圖2(d))、垂直于穴位(見圖2(e))吸附時的吸附能較高,而在頂部鐵位(見圖2(b))、平行于Fe—S鍵(見圖2(c))和平躺在穴位(見圖2(f))吸附時的吸附能較低,且以平躺在穴位上以一個氧原子對著頂部硫原子、另一個氧原子對著表面鐵原子(見圖 2(g))時的吸附能最低,表明這種吸附方式最為穩定;在方鉛礦表面,除氧分子以平躺于穴位且兩個氧原子分別對著兩個硫原子(見圖3(f))吸附在表面時的吸附能最低,吸附最穩定外,其他吸附方式:頂部硫位(見圖 3(a))、頂部鉛位(見圖3(b))、平行于S—Pb鍵(見圖3(c))、平躺在穴位(處于鉛原子之間)(見圖 3(d))以及垂直于穴位(見圖 3(e))的氧分子吸附能都較高。

表2 方鉛礦表面原子配位及位移Table 2 Atomic coordination and displacements of galena surface

圖2 氧分子在黃鐵礦(100)面不同位置的平衡吸附構型Fig. 2 Equilibrium adsorption of O2 on different sites of pyrite (100) surface (Numbers shown near bond indicating bond length in nm. Arrows are indicators of x, y and z axes): (a) Top S site; (b) Top Fe site; (c) Parallel to Fe—S bond; (d) Parallel to S—S bond;(e) Vertical to hollow; (f) Parallel to hollow (between Fe atoms); (g) Parallel to hollow (between Fe and S atoms)
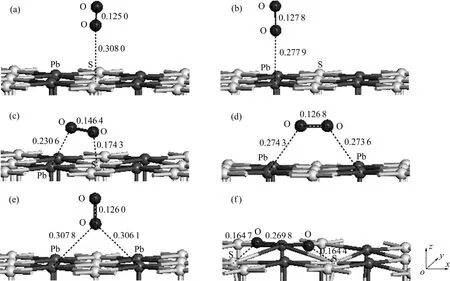
圖3 氧分子在方鉛礦(100)面不同位置的平衡吸附構型Fig. 3 Equilibrium adsorption of O2 on different sites of galena (100) surface (Numbers shown near bond indicating bond length in nm. Arrows are indicators of x, y and z axes): (a) Top S site; (b) Top Pb site; (c) Parallel to Pb—S bond; (d) Parallel to hollow(between Pb atoms); (e) Vertical to hollow; (f) Parallel to hollow (between S atoms)

表3 氧分子在黃鐵礦(100)面的吸附能Table 3 Adsorption energy of O2 on pyrite (100) surface

表4 氧分子在方鉛礦(100)面吸附時的吸附能Table 4 Adsorption energy of O2 on galena (100) surface
在黃鐵礦和方鉛礦表面吸附后的氧分子都發生了解離,并分別與表面的原子成鍵。從氧分子在兩種礦物表面上的最穩定吸附方式可以知道,在黃鐵礦表面,氧原子分別與硫和鐵原子鍵合,而在方鉛礦表面,氧原子只與硫原子鍵合而未與鉛原子鍵合。氧分子在黃鐵礦和方鉛礦表面的吸附能分別為-2.522 eV(見圖2(g))和-1.191 eV(見圖 3(f)),前者明顯低于后者,表明其與黃鐵礦表面的相互作用更強,在黃鐵礦表面的反應活性更高,這也體現在不同表面吸附后的 O—O鍵長和O—S鍵長的區別中。在黃鐵礦和方鉛礦表面上,O—O鍵長分別為0.284 2和0.269 8 nm,氧分子在黃鐵礦表面的解離更徹底;O—S鍵長分別為0.149 6和0.164 4 nm,氧原子與黃鐵礦表面的硫原子之間的鍵合更為緊密。從以上的分析可以知道,當氧分子吸附后,黃鐵礦表面上的硫原子被氧化得更為徹底,即所帶正價將更高,這與實際情況相符,即黃鐵礦的陽極氧化產物主要硫組分為硫酸鹽(S O),而方鉛礦的陽極氧化產物主要硫組分為元素硫(S0)[3]。這也表明黃鐵礦具有較差的無捕收劑浮選特性,而方鉛礦具有較好的無捕收劑浮選行為。
2.3 表面原子電荷分析
圖4所示為黃鐵礦和方鉛礦表面原子的Mulliken電荷,原子上的數字表示電荷值,單位為 e,圖為頂視圖。黃鐵礦(100)表面硫二聚體中的 S1原子位于表面頂部,S2原子位于表面底部(見圖4(a))。從圖4(a)可以看出,S1原子帶電荷-0.10 e,而S2原子帶電荷-0.02 e。另外,與鐵原子配位不同方向上的硫原子所帶電荷不同,處于表面底部的硫原子(S2和S3)所帶負電荷少于表面頂部的硫原子(S4和S5)。表面鐵原子帶正電荷0.08 e。從氧分子吸附后的表面原子電荷(見圖4(b))可以看出,與氧成鍵的 S1和Fe1原子失去了較多的電荷給氧原子而分別帶正電荷0.74 e和0.36 e,而與S1成鍵的O1原子所帶負電荷(-0.76 e)遠多于與Fe1原子成鍵的O2原子(-0.44 e),表明氧原子從表面硫原子上獲得的電荷多于從鐵原子上獲得的電荷。另外,除與表面頂部 S1原子配位的硫原子和其余鐵原子(除Fe1)得到少量電荷外,氧分子周圍的其余硫原子則失去少量電荷。氧分子吸附對更遠處的表面原子的電荷影響較小。

圖4 氧分子吸附前后氧原子及表面原子的Mulliken電荷Fig. 4 Mulliken charges of oxygen atom and surface atoms before and after O2 adsorption (Numbers on atom indicating atomic charge in e. Arrows indicating x and y axes): (a) Pyrite surface before O2 adsorption; (b) Pyrite surface after O2 adsorption; (c) Galena surface before O2 adsorption; (d) Galena surface after O2 adsorption
在理想的方鉛礦(100)表面上,鉛原子帶正電荷0.61 e而硫原子帶負電荷-0.68 e(見圖4(c)),氧分子吸附后對其周圍原子的電荷影響較為明顯。分別與氧原子成鍵的S1和S2原子所帶電荷已從原來的負電荷到吸附氧后略帶正電荷(0.07 e),氧對距離稍遠的硫原子電荷影響很小,靠近氧原子的鉛原子(Pb1和Pb2)失去電子,而離氧較遠的鉛原子(Pb3和Pb4)則得到極少量的電子,由原來的0.61 e變為0.58 e。另外,氧分子吸附對表面硫原子的構型產生了較為明顯的影響,與氧成鍵的S1和S2原子沿著x軸被排斥開來。
從更詳細的原子電荷布居分析可以了解原子之間的電荷轉移情況,表5和表6所列分別為氧分子在黃鐵礦和方鉛礦表面吸附前和吸附后的表面原子及氧原子的電荷布居值。由表5可以知道,氧分子在黃鐵礦表面吸附后,與氧原子成鍵的 S1原子(頂部硫)的 3s軌道失去少量電子而3p軌道都失去較多電子,離氧稍遠處的S2和S3原子的3s軌道電子基本沒有變化,但3p軌道失去非常少量電子;鐵原子(Fe1)的4s軌道電子不變,4p軌道得到非常少量的電子,而3d軌道則失去了較多電子;氧原子的 2s軌道電子沒有發生變化,與S1成鍵的O1原子的2p軌道比與Fe1原子成鍵的O2原子的2p軌道得到更多的電子。由此可知,氧分子與黃鐵礦表面的反應,主要由硫原子的 3p軌道、鐵原子的3d軌道和氧原子的2p軌道參與。
由表6可以看出,氧分子在方鉛礦表面吸附后,與氧成鍵的硫原子(S2)的3s軌道失去少量電子,而3p軌道則失去大量的電子;氧分子周圍的鉛原子(Pb1和Pb2)的 6s軌道電子基本沒有變化,6p軌道失去較多電子;離氧吸附位置稍遠的鉛原子(Pb3)的 6s軌道電子基本沒有變化,而6p軌道則得到少量電子。此外,鉛原子的5d軌道電子數沒有變化,表明5d軌道電子沒有參與氧氣的反應。氧的2s軌道得到少量電子,而2p軌道得到較多的電子。由此可知,氧分子與方鉛礦表面的反應,主要由硫原子的3p軌道、鉛原子的6p軌道和氧原子的2p軌道參與。
從圖5的電荷差分密度(見圖5(a)和(b),黑色區域表示電子富集,白色區域表示電子缺失,背景色表示電子密度為零)和電荷密度圖(見圖 5(c)和(d))可以看到,吸附后氧原子周圍電子富集而與之配位的鐵原子和硫原子周圍則呈電子缺失狀態,氧與礦物表面的原子發生相互作用而成鍵,而由于在表面解離成單氧狀態,氧原子之間已經不成鍵。
2.4 氧分子吸附對黃鐵礦和方鉛礦表面態的影響
從前面的原子電荷布居分析可知:氧分子在黃鐵礦和方鉛礦表面吸附時,主要是氧原子、硫原子和鉛原子的p軌道以及鐵原子的d軌道參與相互作用,因此,圖6和圖7所示分別為氧分子吸附前后黃鐵礦和方鉛礦表面及表面主要參與反應的原子態密度,并考察參與反應的主要電子軌道態密度變化。氧的外層 p電子組態為:本計算結果與實際非常一致。如圖所示,在-6和-4.5 eV處有一個分別由成鍵σ2pz態和π2px(π2py)組成的態密度峰(px和py的原子軌道態密度曲線是重合的);費米能級處的態密度則由半滿的反鍵)態組成;最后,在約1.8 eV處存在一個空反鍵態。
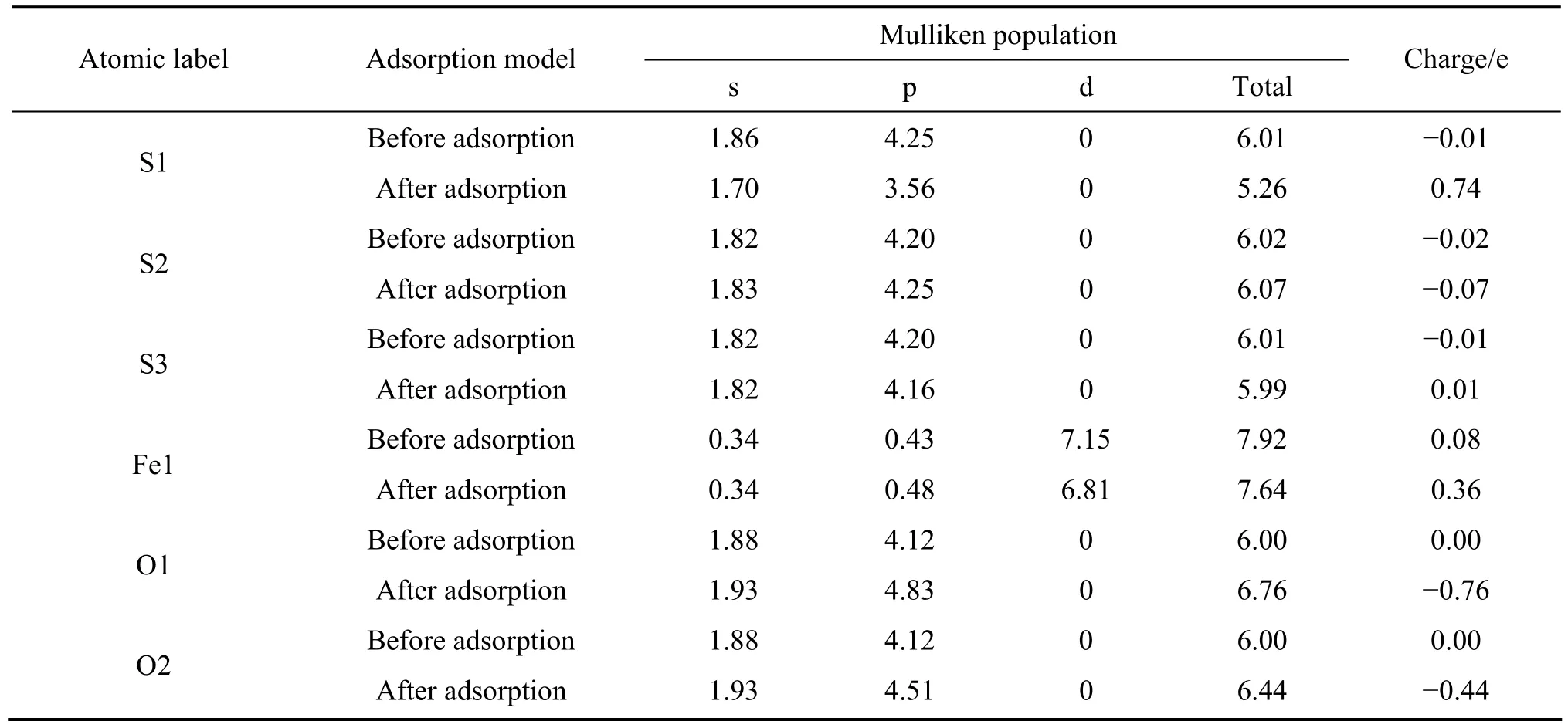
表5 氧分子吸附前后黃鐵礦表面原子及氧原子的Mulliken電荷布居Table 5 Mulliken charge populations of O atom and surface atom before and after O2 adsorption on pyrite surface

表6 氧分子吸附前后方鉛礦表面原子及氧原子的Mulliken電荷布居Table 6 Mulliken charge populations of O atom and surface atom before and after O2 adsorption on galena surface

圖5 氧分子吸附后黃鐵礦和方鉛礦的電荷密度和差分電荷密度Fig. 5 Electron density and electron density difference maps of pyrite and galena after O2 adsorption: (a) Electron density difference of pyrite; (b) Electron density difference of galena; (c) electron density of pyrite; (d) Electron density of galena
在黃鐵礦表面,與氧成鍵的硫原子和鐵原子的態密度發生了極為明顯的變化,而氧氣分子本身的態密度也發生了變化,說明氧吸附對礦物表面態產生了明顯的影響。本文作者主要通過討論占據電子態來討論氧與表面的相互作用。費米能級以下-8~0 eV能量范圍內吸附氧的2p軌道電子態密度呈連續分布狀態,電子非局域性增強;吸附氧后的 S1原子在費米能級以下-8~-1.50 eV范圍內的3p電子態密度峰向低能方向移動,而-1.5~0 eV的3p電子態密度明顯降低;Fe1原子的3d軌道電子對態密度的貢獻占主導地位,吸附氧后在-6~0 eV范圍內形成連續分布,并且-1.5~0 eV范圍內的 3d電子態密度明顯降低。氧吸附對方鉛礦表面也產生了明顯影響。在費米能級以下-6~0 eV范圍內吸附后的氧2p軌道電子態密度呈連續分布狀態,且主要集中在-5.5~3.5 eV能量范圍內;S2原子由于吸附氧導致原來處于-4~0 eV范圍的連續3p電子態在-6~0 eV 能量范圍內形成兩個集中態,即集中在-5~-4 eV和-1~0 eV能量范圍內;Pb1原子的整體態密度向低能方向移動了較小距離且 6p電子態密度明顯降低(5d軌道電子能量非常低,在氧吸附過程中沒有參與反應),-3~-1 eV能量范圍內的6p電子態由于氧吸附而幾乎消失。
由態密度圖可以看出,在黃鐵礦表面,氧與硫成鍵時,電子主要由硫的3p軌道向氧的2p軌道轉移,而與鐵成鍵時,則主要由鐵的3d軌道電子向氧的2p軌道轉移,形成d→p反饋鍵;在方鉛礦表面上,電子主要由硫和鉛的6p軌道向氧的2p軌道轉移,而鉛的5d軌道由于沒有參與反應,未能與氧的2p軌道形成d→p反饋鍵。因氧與鐵之間d→p反饋鍵的形成,氧分子在黃鐵礦表面的吸附將更為穩定,黃鐵礦表面將被氧化得更為徹底,這與前面對吸附能和鍵長的計算的結果一致。
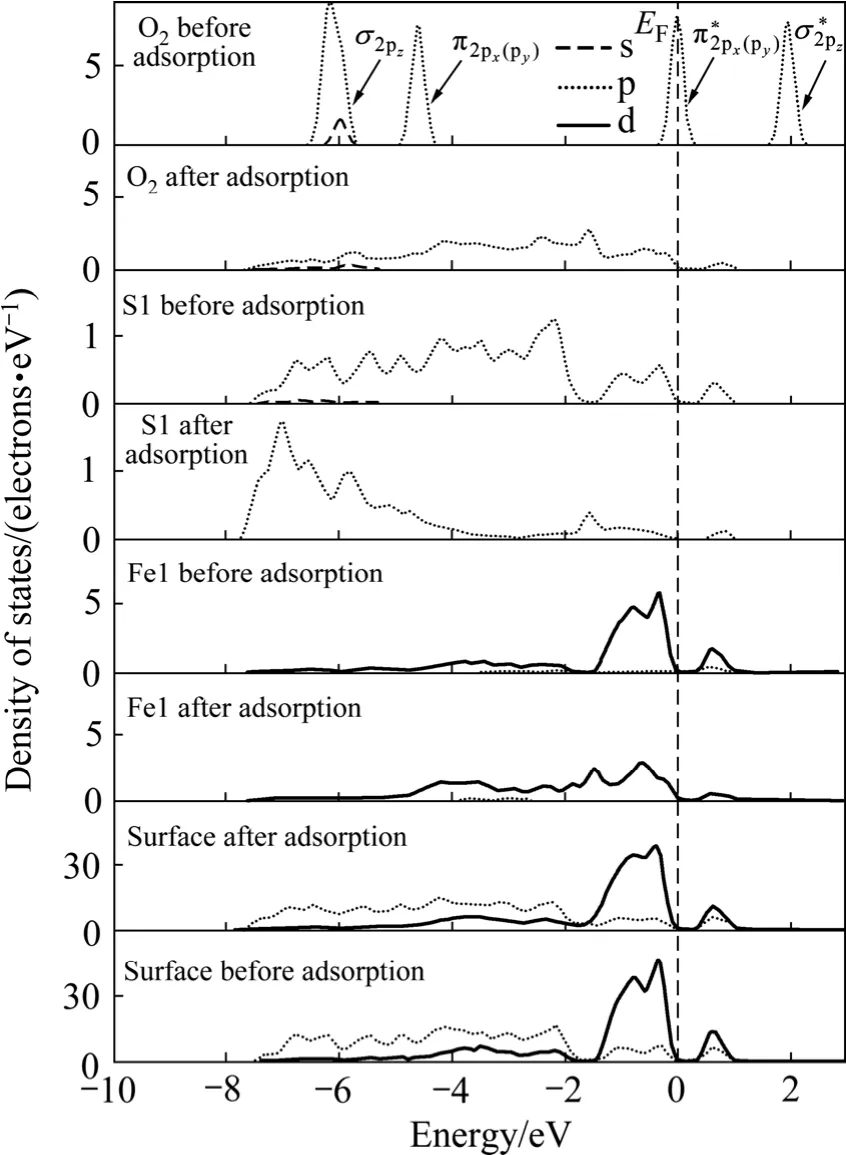
圖6 氧分子吸附前后黃鐵礦表面原子氧氣態密度Fig. 6 Density of states of surface atom and oxygen before and after O2 adsorption on pyrite surface (EF indicates position of Fermi level, and the value is 0 eV)
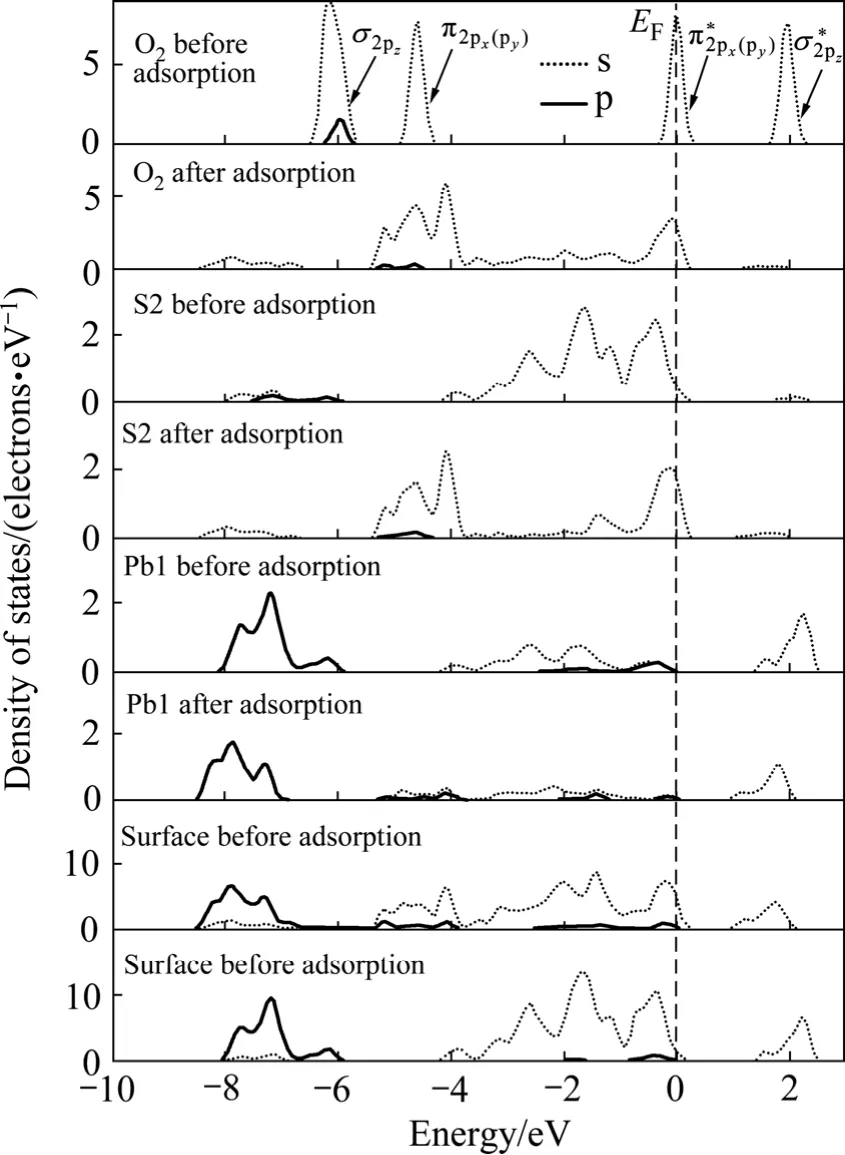
圖7 氧分子吸附前后方鉛礦表面原子和氧氣態密度Fig. 7 Density of states of surface atom and oxygen before and after O2 adsorption on galena surface

圖8 氧分子吸附前后黃鐵礦表面原子和氧原子的自旋態密度Fig. 8 Spin density of states of surface atom and oxygen atom before and after O2 adsorption on pyrite surface

圖9 氧分子吸附前后方鉛礦表面原子和氧原子的自旋態密度Fig. 9 Spin density of states of surface atom and oxygen atom before and after O2 adsorption on galena surface
圖8和9所示分別為氧分子吸附前后的黃鐵礦和方鉛礦表面原子自旋態密度。圖中僅顯示了主要參與反應的軌道電子自旋,即S 3p、O 2p、Fe 3d和Pb 6p軌道電子,主要考察其在費米能級(EF)附近的變化,α和β分別代表向上和向下自旋。由圖8可以看出,氧分子吸附前的鐵原子(Fe1)為低自旋態,吸附后產生了自旋,而與鐵原子成鍵的氧原子(O2)也產生了自旋;吸附前后的硫原子(S1)都呈低自旋態,與硫原子成鍵的氧原子(O1)也為低自旋態。由圖9可以看出,吸附前后氧、硫和鉛原子都呈低自旋態,沒有產生自旋現象。從電子自旋分析可以看出,在黃鐵礦表面上的氧和鐵原子由于發生相互作用而產生了自旋現象,而在方鉛礦表面的氧則沒有發生自旋現象,具有磁性的物質之間更容易產生相互吸引,因此,氧分子在黃鐵礦表面的反應活性將比在方鉛礦表面大。
3 結論
1) 氧分子在黃鐵礦和方鉛礦表面具有明顯不同的吸附方式。氧分子和黃鐵礦作用產生了自旋,導致氧分子在黃鐵礦表面的反應活性更高,而氧分子與方鉛礦表面相互作用后沒有產生自旋,削弱了氧分子在方鉛礦表面的吸附。
2) 在黃鐵礦表面,氧與金屬鐵原子之間形成d→p反饋鍵,而在方鉛礦表面沒有反饋鍵形成,這也增強了氧在黃鐵礦表面的吸附;黃鐵礦表面氧化傾向于生成高價硫,氧化更為徹底,而方鉛礦表面氧化后傾向于形成低價硫,這與實際檢測結果一致,從理論上解釋了黃鐵礦和方鉛礦無捕收劑浮選差異的本質原因。
REFERENCES
[1] PLASKIN I N. Interaction of minerals with gases and reagents in flotation[J]. Mining Engineering, 1959, 214: 319-324.
[2] HOFFMAN I, NATHANIEL A. Flotation[J]. Industrial and Engineering Chemistry, 1957, 49(3): 493-496.
[3] 馮其明, 陳 藎. 硫化礦物浮選電化學[M]. 長沙: 中南工業大學出版社, 1992: 14-90.FENG Qi-ming, CHEN Jin. Electrochemistry of sulfide mineral flotation[M]. Changsha: Central South University of Technology Press, 1992: 14-90.
[4] WOODS R. Electrochemical potential controlling flotation[J].International Journal of Mineral Processing, 2003, 72(1/4):151-162.
[5] WOODS R. Recent advances in electrochemistry of sulfide mineral flotation[J]. Transactions of Nonferrous Metals Society of China, 2000, 10(Special Issue): 26-29.
[6] UHLIG I, SZARGAN R, NESBITT H W, LAAJALEHTO K.Surface states and reactivity of pyrite and marcasite[J]. Applied Surface Science, 2001, 179(1/4): 222-229.
[7] TOSSELL J A, VAUGHAN D J. Electronic structure and the chemical reactivity of the surface of galena[J]. Canadian Mineralogist, 1987, 25: 381-392.
[8] MURPHY R, STRONGIN D R. Surface reactivity of pyrite and related sulfides[J]. Surface Science Reports, 2009, 64(1): 1-45.
[9] MENDIRATTA N K. Kinetic studies of sulfide mineral oxidation and xanthate adsorption[D]. Virginia: Virginia Polytechnic Institute and State University, 2000: 43-49.
[10] AHLBERG E, BROO A E. Oxygen reduction at sulphide minerals. 1. A rotating ring disc electrode (RRDE) study at galena and pyrite[J]. International Journal of Mineral Processing,1996, 46(1/2): 73-89.
[11] AHLBERG E, BROO A E. Oxygen reduction at sulphide minerals. 2. A rotating ring disc electrode (RRDE) study at galena and pyrite in the presence of xanthate[J]. International Journal of Mineral Processing, 1996, 47(1/2): 33-47.
[12] AHLBERG E, BROO A E. Oxygen reduction at sulphide minerals. 3. The effect of surface pre-treatment on the oxygen reduction at pyrite[J]. International Journal of Mineral Processing, 1996, 47(1/2): 49-60.
[13] HAUNG H H, MILLER J D. Kinetics and thermochemistry of amyl xanthate adsorption by pyrite and marcasite[J].International Journal of Mineral Processing, 1978, 5(3):241-266.
[14] PILLAI K C, BOCKRIS J O'M. A quantitative examination of the mixed potential mechanism in mineral flotation[J]. Journal of the Electrochemical Society, 1984, 131(3): 568-579.
[15] BECKER U, HOCHELLA M F Jr. The calculation of STM images, STS spectra, and XPS peak shifts for galena: New tools for understanding mineral surface chemistry[J]. Geochimica et Cosmochimica Acta, 1996, 60(13): 2413-2426.
[16] RAIKAR G N, THURGATE S M. An Auger and EELS study of oxygen adsorption on FeS2[J]. Journal of Physics: Condensed Matter, 1991, 3(12): 1931-1939.
[17] ROSSO K M, BECKER U, HOCHELLA M F. The interaction of pyrite {100} surfaces with O2and H2O: Fundamental oxidation mechanisms[J]. American Mineralogist, 1999, 84(10):1549-1561.
[18] KENDELEWICZ T, DOYLE C S, BOSTICK B C, BROWN G E. Initial oxidation of fractured surfaces of FeS2(100) by molecular oxygen, water vapor, and air[J]. Surface Science, 2004,558(1/3): 80-88.
[19] 王淀佐, 龍翔云, 孫水裕. 硫化礦的氧化與浮選機理的量子化學研究[J]. 中國有色金屬學報, 1991, 1(1): 15-23.WANG Dian-zuo, LONG Xiang-yun, SUN Shui-yu. Quantum chemical mechanism on the surface oxidation and flotation of sulfide minerals[J]. The Chinese Journal of Nonferrous Metals,1991, 1(1): 15-23.
[20] SUN W, HU Y H, QIU G Z, QIN W Q. Oxygen adsorption on pyrite (100) surface by density functional theory[J]. Journal of Central South University of Technology, 2004, 11(5): 385-390.
[21] CLARK S J, SEGALL M D, PICKARD C J, HASNIP P J,PROBERT M I J, REFSON K, PAYNE M C. First principles methods using CASTEP[J]. Zeitschrift fur Kristallographie, 2005,220(5/6): 567-570.
[22] SEGALL M D, LINDAN P J D, PROBERT M J, PICKARD C J,HASNIP P J, CLARK S J, PAYNE M C. First-principles simulation: Ideas, illustrations and the CASTEP code[J]. Journal of Physics: Condensed Matter, 2002, 14(11): 2717-2744.
[23] VANDERBILT D. Soft self-consistent pseudopotentials in a generalized eigen value formalism[J]. Physical Review B, 1990,41(11): 7892-7895.
[24] MONKHORST H J, PACK J D. Special points for Brillouin-zone integrations[J]. Physical Review B, 1976, 13(12):5188-5192.
[25] PACK J D, MONKHORST H J. Special points for Brilliouin-zone zntegrations—A reply[J]. Physical Review B,1977, 16(4): 1748-1749.
[26] PRINCE K C, MATTEUCCI M, KUEPPER K. CHIUZBAIAN S G, BARKOWSKI S, NEUMANN M. Core-level spectroscopic study of FeO and FeS2[J]. Physical Review, 2005, 71(8):085102-1-085102-9.
[27] WYCKOFF R W G. Crystal structures [M]. New York:Interscience Publishers, 1963: 85-237.
[28] ROSSO K M, BECKER U, HOCHELLA M F Jr. Atomically resolved electronic structure of pyrite (100) surfaces: An experimental and theoretical investigation with implications for reactivity[J]. American Mineralogist, 1999, 84(10): 1535-1548.
[29] CHATURVERDI S, KATZ R, GUEVREMONT J, SCHOONER M A A, STROGIN D R. XPS and LEED study of a naturally occurring single crystal of pyrite[J]. American Mineralogist,1996, 81(1/2): 261-264.
[30] HUNG A, MUSCAT J, YAROVSDY I, RUSSO S P.Density-functional theory studies of pyrite FeS2(100) and (110)surfaces[J]. Surface Science, 2002, 513(3): 511-524.
[31] 李炳瑞. 結構化學[M]. 北京: 高等教育出版社, 2004: 85-86.LI Bing-rui. Structural chemistry[M]. Beijing: Higher Education Press, 2004: 85-86.
Adsorption of O2on pyrite and galena surfaces
LI Yu-qiong1, CHEN Jian-hua2, LAN Li-hong1,3, GUO Jin4
(1. School of Chemistry and Chemical Engineering, Guangxi University, Nanning 530004, China;2. College of Resources and Metallurgy, Guangxi University, Nanning 530004, China;3. College of Chemistry and Ecoengineering, Guangxi University for Nationalities, Nanning 530006, China;4. College of Physics Science and Engineering, Guangxi University, Nanning 530004, China)
The adsorption of oxygen molecule (O2) on the pyrite and galena surfaces was studied using density functional theory (DFT). The calculated results show that the surface relaxation of pyrite and galena is small. O2dissociates after adsorption on the pyrite and galena surfaces, and the adsorption energy of O2on pyrite is much lower than that on the galena. On the pyrite surface, oxygen atom (O) bonds with sulfur (S) and iron (Fe) atoms and the electrons are transferred from Fe and S atoms to O, and the reactions are mainly S 3p, O 2p and Fe 3d states involved, forming the d→p back bonding between Fe and O. While on the galena surface, oxygen atom only bonds with sulfur atom, and the reactions are mainly S 3p, O 2p and Pb 6p states involved, without forming d→p back bonding. The bonded Fe and O atoms are spin-polarized after the adsorption of O2, while the galena surface atoms and adsorbed O atom are still low-spin states.
pyrite; galena; O2adsorption; density functional theory
TD923
A
1004-0609(2012)04-1184-11
國家自然科學基金資助項目(50864001)
2011-01-20;
2011-07-11
陳建華,教授,博士;電話:0771-3232200;E-mail: jhchen1971@sina.com
(編輯 龍懷中)
