碳酸鹽共沉淀法制備Li[Li0.2Co0.13Ni0.13Mn0.54]O2中加料方式對產物性能的影響
2012-11-09 10:42:36趙軍峰王偉剛曹雁冰胡國榮彭忠東
無機化學學報 2012年1期
杜 柯 趙軍峰 王偉剛 黃 霞 曹雁冰 胡國榮 彭忠東
(中南大學冶金科學與工程學院輕金屬及工業電化學研究所,長沙 410083)
碳酸鹽共沉淀法制備Li[Li0.2Co0.13Ni0.13Mn0.54]O2中加料方式對產物性能的影響
杜 柯*趙軍峰 王偉剛 黃 霞 曹雁冰 胡國榮 彭忠東
(中南大學冶金科學與工程學院輕金屬及工業電化學研究所,長沙 410083)
采用碳酸鈉和碳酸氫銨作為沉淀劑和絡合劑,在水溶液中共沉淀Mn2+、Ni2+和Co2+以獲得混合過渡金屬元素的碳酸鹽沉淀前驅體Mn0.675Ni0.1625Co0.1625CO3。并進一步合成高容量鋰離子電池正極材料Li[Li0.2Co0.13Ni0.13Mn0.54]O2。考察了3種不同加料方式對共沉淀前驅體的結構、形貌和元素比例的影響,以及對最終產物的結構、形貌和電化學性能的影響。
鋰離子電池;Li[Li0.2Co0.13Ni0.13Mn0.54]O2;正極材料;共沉淀
近年來,yLi2MnO3·(1-y)Li[NixCo1-2xMnx]O2型固溶體[1-5]正極材料因其具有比容量高、價格低廉、循環穩定、對環境友好等優點吸引了國內外專家學者的深入研究[6-11],被認為是一種很有發展前景的下一代鋰離子電池正極材料。其中綜合性能較佳的Li [Li0.2Co0.13Ni0.13Mn0.54]O2也即 0.6Li2MnO3·0.4LiCo1/3Ni1/3Mn1/3O2,首次放電比容量可高達250 mAh·g-1。同時,經過適當的包覆改性可使材料具有較好的循環穩定性[12-13]。
Li[Li0.2Co0.13Ni0.13Mn0.54]O2目前制備方法主要有固相法、溶膠凝膠法、共沉淀法等。其中共沉淀法可以克服普通固相法混合不均勻的缺點,使原料在原子水平上混合,從而得到微觀分布均勻的材料,因此被認為是制備混合過渡金屬元素材料較好的方法。傳統Li[NixCo1-2xMnx]O2系列三元材料一般是先制備氫氧化物前驅體,再與Li2CO3或者LiOH·H2O等不同鋰源混合熱處理。但是由于Mn(OH)2不穩定,很容易被氧化為三價的 MnOOH,甚至氧化為四價的MnO2,造成前驅體組分不均勻,導致在熱 處理過程中形成雜相,影響合成材料的電化學性能[14-15]。Li[Li0.2Co0.13Ni0.13Mn0.54]O2材料由于具有較高的錳含量,所以采用氫氧化物共沉淀,會使錳的氧化問題更嚴重。而以碳酸鹽共沉淀技術制備前驅體,可使前驅體中金屬離子全部以穩定的碳酸鹽形式存在,同時反應的pH值一般較氫氧化物共沉淀低,球形形貌易于控制,合成的最終產物具有較高的放電容量[16-17]。
本文針從Mn、Ni、Co的硫酸鹽水溶液制備碳酸鹽沉淀前驅體的實驗過程,進行了3種不同加料方式的研究。考察了它們對合成的前驅體在結構、形貌和化學組成上的影響,以及對進一步合成的電極活性材料的結構、形貌和電化學性能的影響。
1 實驗部分
1.1 材料的制備
如圖 1所示,以 NiSO4·6H2O、CoSO4·7H2O、MnSO4·H2O為原料,按照產物中過渡金屬元素的計量比配制金屬離子總濃度為2 mol·L-1的水溶液,同時配制 2 mol·L-1的 Na2CO3溶液及 2 mol·L-1的NH4HCO3溶液。分別將3種溶液以不同加料方式加入高速攪拌的反應器中:(A)Na2CO3溶液作為反應器中底液,金屬離子溶液以一定速度滴入反應器中;(B)以一定量的去離子水為底液,Na2CO3溶液與金屬離子溶液同時以一定速度并流加入反應器中;(C)以一定量的去離子水為底液,Na2CO3溶液、NH4HCO3溶液、金屬離子溶液三液并流加入反應器中。反應得到沉淀Mn0.675Ni0.1625Co0.1625CO3。用去離子水抽濾洗滌沉淀至無硫酸根殘留 (用BaCl2溶液檢測),在120℃真空干燥箱中干燥10 h,粉碎過篩得到Mn0.675Ni0.1625Co0.1625CO3前驅體。測試所得前驅體中鎳鈷錳含量,按計量比混合Li2CO3,在乙醇介質中球磨2 h,烘干過篩,然后在馬弗爐中加熱至900℃,保溫10 h,隨爐冷卻,粉碎過篩,得到Li[Li0.2Co0.13Ni0.13Mn0.54]O2材料。
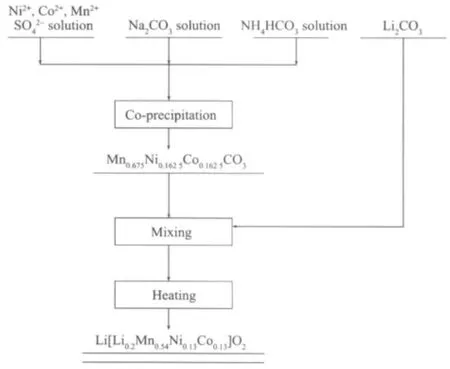
圖1 碳酸鹽共沉淀制備Li[Li0.2Mn0.54Ni0.13Co0.13]O2的流程圖Fig.1 Flow chart of preparing Li[Li0.2Co0.13Ni0.13Mn0.54]O2by carbonate co-precipitation method
1.2 結構和形貌的表征
采用日本Rigaku公司生產Minflex型的X射線自動衍射儀對試樣進行物相鑒定。測試條件為:Cu Kα1射線,管電壓、管電流分別為40 kV和200 mA,掃描范圍為5°~80°。
采用日本JEOL公司JSM-5600LV型掃描電子顯微鏡對合成目標產物的顆粒大小和表面形貌進行觀察。
鎳鈷含量采用EDTA配位滴定分析,錳含量采用硫酸亞鐵銨氧化還原滴定分析。
1.3 電化學性能測試
將材料組裝成CR2025型扣式電池進行充放電循環測試。采用涂膜法制備電極,以N-甲基-2-吡咯烷酮(NMP)為溶劑,按質量比8∶1∶1分別稱取正極材料、乙炔黑和PVDF,混合均勻后,涂在預處理過的鋁箔上,放入真空干燥箱中在120℃干燥得到正極片。純金屬鋰片作負極,Celgard 2400多孔聚乙烯膜為隔膜,1 mol·L-1LiPF6的EC、DMC和EMC(1∶1∶1,V/V/V)的混合溶液作為電解液,在水和氧含量均小于1 mL·m-3的手套箱中組裝成模擬電池。在室溫下用LAND電池測試系統對模型電池進行恒電流循環充放電測試,電壓范圍為2~4.8V,電流密度1C=300 mA·g-1。
2 結果與討論
2.1 共沉淀加料方式對合成前驅體影響
2.1.1 不同加料方式對前驅體形貌及粒度分布的影響
圖2為3種不同加料方式合成的前驅體Mn0.675Ni0.1625Co0.1625CO3在 2個不同放大倍率下的SEM圖。可以看出,將金屬離子溶液直接滴入Na2CO3溶液的加料方式(A)所合成的前驅體一次顆粒較小,團聚成的顆粒形貌不規則,大小不均勻。而Na2CO3溶液與金屬離子溶液并流加料方式 (B)和Na2CO3溶液、NH4HCO3溶液、金屬離子溶液三液并流的加料方式(C)所合成的前驅體均呈現類球形,其中C加料方式獲得的前驅體團聚粒子分布更均勻。

A/A′:Feeding way A;B/B′:Feeding way B;C/C′:Feeding way C圖2 不同加料方式合成前驅體的SEM圖Fig.2 SEM images of precursors prepared by different raw materials feeding ways
圖3為不同加料方式合成前驅體的粒度分布圖,表1為對應的參數。可以看到,加料方式(A)所合成的前驅體顆粒具有最小的D50值(16.20 μm),但是粒徑分布范圍較寬。這是因為Na2CO3做底液,金屬離子滴入溶液中時,沉淀劑始終是過量的,溶液體系具有較大的過飽和度,新加入的金屬離子能夠快速形成新的晶核,晶體顆粒長大速度較慢,所以合成的前驅體顆粒較小。同時沒有絡合劑的存在,沒有絡合反應與沉淀反應的競爭,顆粒再溶解再生長過程較慢,所以顆粒分布不規整。并流加料方式(B)所合成的前驅體D50值為18.18 μm粒度分布峰較窄。這是因為在并流加料開始,沉淀劑與金屬離子會形成大量晶核,在繼續加入金屬離子及沉淀劑時,在攪拌作用下金屬離子和沉淀劑迅速分散在溶液中,反應體系中沉淀劑與金屬離子濃度都較低,溶液中過飽和度較小,在形成新的晶核的同時,會伴隨晶體顆粒逐漸長大,所以Na2CO3與金屬離子并流加料得到的前驅體相對粒徑較大,且分布較為均勻。三液并流的加料方式(C)所得到的前驅體,具有最大的D50值(19.64 μm),但是粒度分布最均勻,粒度分布峰最窄。這是因為該過程與B過程同樣使得溶液中具有較小的過飽和度,有利于晶核的形成及長大,同時加入的配位劑不但可以保證金屬元素均勻共沉淀,而且使沉淀反應與配位反應競爭進行,可以優化沉淀的再溶解及長大過程,使得材料具有球形形貌,且分布均勻。
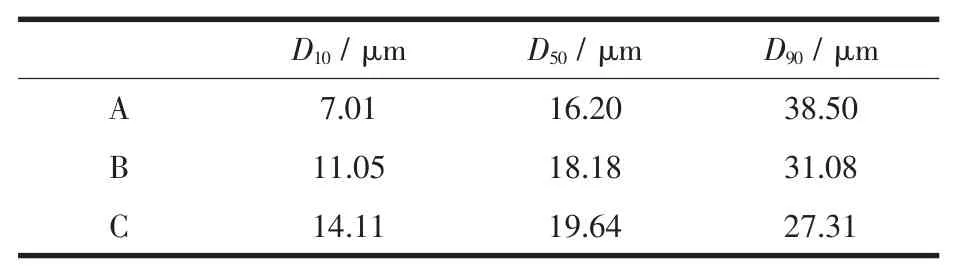
表1 不同加料方式合成前驅體的粒度參數Table 1 Size parameters of precursor particles prepared by different raw materials feeding ways
2.1.2 不同加料方式對前驅體化學成分的影響
前驅體的化學成分對材料性能影響巨大,符合化學計量比的前驅體則是合成化學計量比材料的前提。由于實驗中3種金屬離子的碳酸鹽溶度積常數有一定差別,不同的加料方式,使得反應體系中金屬離子與沉淀劑濃度有差別,所以不同加料方式導致反應過程中不同金屬離子沉淀完全程度不同,導致前驅體中各金屬離子化學成分產生差別。表2是不同加料方式合成前驅體中的過渡元素的組成表,可以看出,3種加料方式得到前驅體中金屬離子摩爾比與理論值都有所偏差。其中A和B實驗中,鈷含量偏高,鎳、錳含量偏低,這是因為3種碳酸鹽的溶度積中:Ksp(CoCO3)=1.4×10-13,Ksp(NiCO3)=6.6× 10-9,Ksp(MnCO3)=1.8×10-11,碳酸鈷最小,在沒有絡合劑存在的情況下會優先沉淀,沉淀較為完全;鎳、錳離子由于溶度積常數較大可能有少量在濾液中損失。C加料方式由于有絡合劑的存在,明顯優化了3種金屬離子的共沉淀過程,金屬離子摩爾比與理論值差別最小。
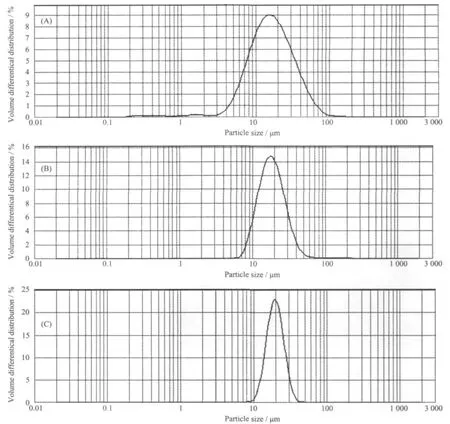
圖3 不同加料方式合成前驅體的粒度分布圖Fig.3 Distribution of precursor particles prepared by different raw materials feeding ways

表2 不同加料方式合成前驅體元素組成Table 2 Elementary analysis of precursor prepared by different raw materials feeding ways
2.1.3 不同加料方式合成前驅體的結構
因為碳酸鹽沉淀體系中不可避免的有氫氧根的存在,一般的碳酸鹽沉淀很容易混雜有堿式碳酸鹽或者氫氧化物沉淀,特別是鎳、鈷含量偏高的情況下更容易得到堿式碳酸鹽。堿式碳酸鹽在前驅體中單獨成相,導致合成材料中雜相的出現,影響材料的電化學性能[18]。從圖4不同加料方式合成前驅體的XRD圖中可以看出,3種方法合成的前驅體XRD圖的主峰都與MnCO3的標準圖相吻合,而與NiCO3、CoCO3的主峰位置有所偏差,主要是因為材料中錳含量較高,而鎳、鈷含量相對較低。且一般認為MnCO3易于形成較好的結晶結構,而NiCO3、CoCO3的結晶度相對較低。從圖4中可以看出3種前驅體中基本都無雜峰,說明3種沉淀方式都得到了碳酸鹽結構的前驅體。其中A加料方式合成的前驅體峰形有一定程度的寬化,可能是A加料方式中錳含量偏離化學計量較多導致的。C加料方式得到前驅體的主峰最尖銳,半峰寬最窄,說明C加料方式得到的前驅體結晶度最高,出現堿式碳酸鹽及氫氧化物的程度更小,更有利于減少合成材料中雜相的出現。
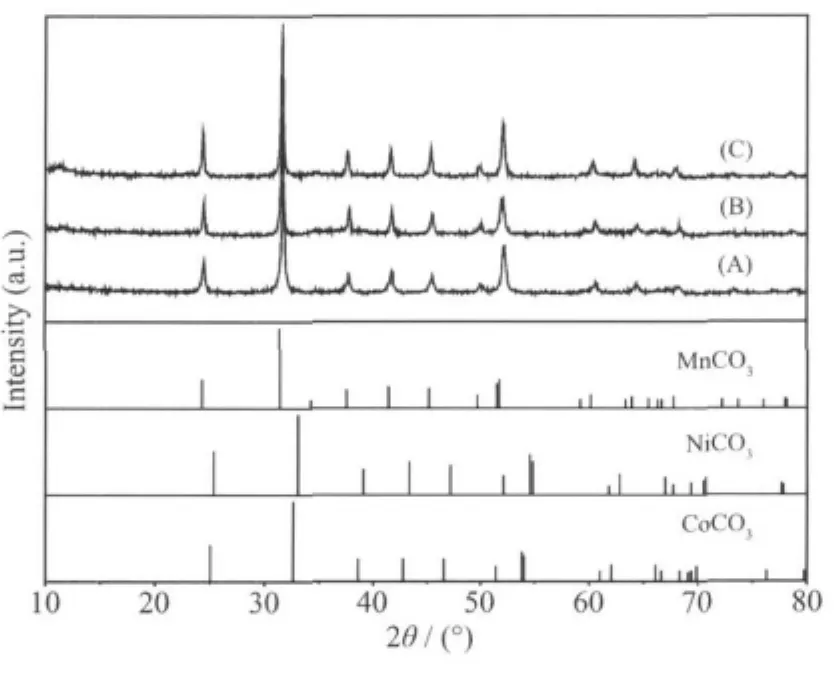
圖4 不同加料方式合成前驅體的XRD圖Fig.4 XRD patterns of precursors prepared by different raw materials feeding ways
2.2 共沉淀加料方式對合成材料的影響
2.2.1 不同加料方式合成前驅體制備材料的形貌
圖 5是不同加料方式合成前驅體制備 Li [Li0.2Co0.13Ni0.13Mn0.54]O2材料的SEM圖。從中可以看出,A加料方式得到前驅體合成的材料雖然顆粒細小,但是晶體形成不完整,顆粒沒有明顯的邊界。B加料方式得到前驅體合成的材料與A加料方式差別不大,同時有部分大顆粒存在。C加料方式得到前驅體合成材料顆粒在1~3 μm,且邊界清晰,晶體形成較好,顆粒分布也比較均勻。

圖5 不同前驅體制備的Li[Li0.2Co0.13Ni0.13Mn0.54]O2材料的SEM圖Fig.5 SEM images of Li[Li0.2Co0.13Ni0.13Mn0.54]O2synthesized by different precursors
2.2.2 不同加料方式合成前驅體制備材料的結構
圖 6是用不同加料方式合成前驅體所制備的 Li[Li0.2Co0.13Ni0.13Mn0.54]O2材料的 XRD圖。 因為Li2MnO3與LiCo1/3Ni1/3Mn1/3O2具有較好的結構相容性,復合材料的XRD主峰主要以層狀結構特征為主,從圖6可以看出,3種加料方式合成前驅體所制備的 Li[Li0.2Co0.13Ni0.13Mn0.54]O2材料都具有單相 α-NaFeO2層狀結構,空間群為R3m,文獻認為(006)/ (102)和(018)/(110)兩組峰分裂越明顯層狀結構越完整[19]。圖6中標示處21°~25°之間的兩個弱峰是R3m空間群所不具有的,文獻認為與Li2MnO3結構特征的C2/m對稱性有關,也就是鋰離子進入過渡金屬層形成LiMn6結構的標志[20]。在此種材料中兩個弱峰的出現也表明形成了良好的Li2MnO3復合材料。從圖6中可以看出,A加料方式合成前驅體所制備的Li[Li0.2Co0.13Ni0.13Mn0.54]O2材料在21°~25°之間的兩個弱峰不明顯,這與錳含量較低,不利于形成Li2MnO3有關。C加料方式合成前驅體所制備的材料的兩個弱峰最明顯,(006)/(102)和(018)/(110)兩組峰分裂也最明顯,即該材料形成了最好的層狀固溶體結構。
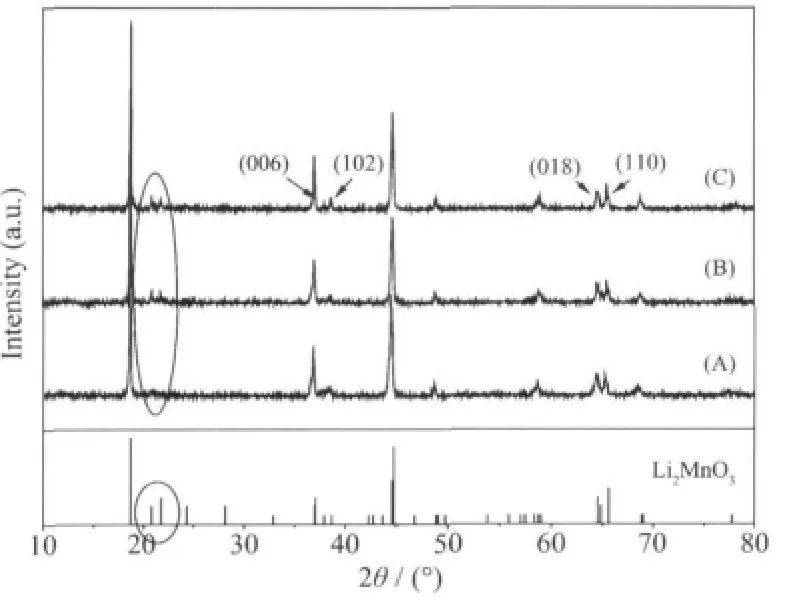
圖6 不同前驅體制備Li[Li0.2Co0.13Ni0.13Mn0.54]O2材料的XRD圖Fig.6 XRD patterns of Li[Li0.2Co0.13Ni0.13Mn0.54]O2 synthesized by different precursors
2.2.3 不同加料方式合成前驅體制備材料的電化學性能
圖7為用3種加料方式獲得的前驅體所制備的Li[Li0.2Co0.13Ni0.13Mn0.54]O2材料的首次充放電曲線圖。可以看出,3種實驗熱處理得到材料的首次充電曲線都呈現出典型的富錳固溶體材料首次充放電曲線:即在充電電壓小于4.5 V時出現S型曲線,是在充電過程中材料脫出鋰的同時,復合材料中LiNi1/3Co1/3Mn1/3O2部分中鎳離子參與電子轉移的固溶體反應。充電電壓大于4.5 V時出現接近L型的充電平臺,主要與復合材料中Li2MnO3部分參與反應有關,即Li2MnO3的高電位電化學活化區,該階段材料脫出鋰離子的同時電子補償由氧離子參與,伴隨有氧氣的放出及新相的生成,所以出現充電平臺[21]。3種前驅體制備的材料0.1C首次放電比容量分別為217.2、226.6、238.4 mAh·g-1。C實驗具有最高的放電比容量,充電平臺較低,放電平臺較高,說明在充放電過程中極化較小。
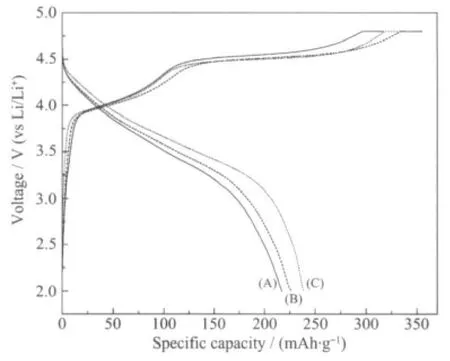
圖7 不同前驅體制備的Li[Li0.2Co0.13Ni0.13Mn0.54]O2材料的0.1C首次充放電曲線Fig.7 Initial charge-discharge curves at 0.1C of Li[Li0.2Co0.13Ni0.13Mn0.54]O2synthesized by different precursors
圖8是用3種加料方式獲得的前驅體所制備的Li[Li0.2Co0.13Ni0.13Mn0.54]O2材料的循環性能圖。A實驗制備的材料經過20次循環后,容量保持率為94%;而B和C實驗獲得的材料都表現出較好的循環性能,50次循環后容量保持率為 98.2%和99.2%。這主要由于實驗A合成的前驅體中金屬離子摩爾比偏離化學計量比較大,熱處理制備材料結晶性能不好,特征的超晶格峰不明顯,層狀結構不完整。同時前驅體顆粒及制備的材料分布都不均勻,充放電過程中,小顆粒與電解液接觸面積大,形成較厚的SEI膜,造成較大的首次不可逆容量及阻礙鋰離子在材料界面的傳輸,導致A實驗得到的材料循環性能不理想。
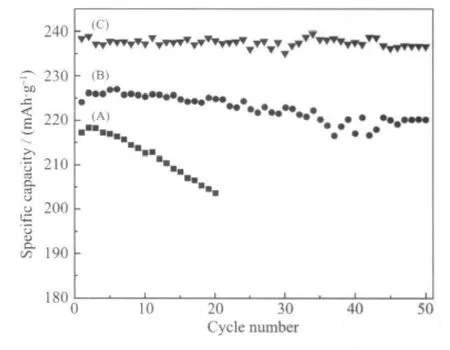
圖8 不同前驅體制備的Li[Li0.2Co0.13Ni0.13Mn0.54]O2材料的0.1C循環曲線Fig.8 Cycle performance of Li[Li0.2Co0.13Ni0.13Mn0.54]O2 synthesized by different precursors
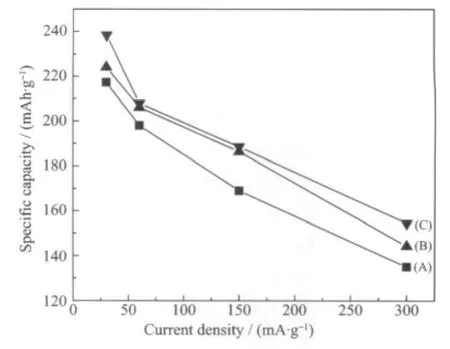
圖9 不同前驅體制備的Li[Li0.2Co0.13Ni0.13Mn0.54]O2材料的倍率性能曲線Fig.9 Rate performance of Li[Li0.2Co0.13Ni0.13Mn0.54]O2 synthesized by different precursors
圖9是用3種加料方式獲得的前驅體所制備的Li[Li0.2Co0.13Ni0.13Mn0.54]O2材料的倍率性能圖。3個樣品的放電容量隨放電電流的增加明顯下降,在300 mA·g-1(1C)的放電電流下,A、B和C實驗獲得的材料的放電比容量分別為135.0、144.3和154.5 mAh· g-1,為0.1C放電比容量的62.1%、64.4%和64.8%。相對而言,C實驗得到的材料表現出較好的倍率性能,這可能主要是由于C實驗合成的前驅體最接近化學計量比,顆粒分布均勻,制備的材料層狀結構完善,在充放電過程中有利于鋰離子的脫嵌。
3 結 論
(1)Na2CO3、NH4HCO3、金屬離子3液并流加料合成的Mn0.675Ni0.1625Co0.1625CO3前驅體顆粒具有較好的球形形貌及合適的粒度分布。
(2)以 3 液 并 流 加 料 合 成 的 Mn0.675Ni0.1625Co0.1625CO3為前驅體所制備的 Li[Li0.2Co0.13Ni0.13Mn0.54] O2材料具有238.4 mAh·g-1的初始放電比容量,50次循環容量保持率在99%以上。
[1]Koyama Y,Makimura Y,Tanaka I,et al.J.Electrochem.Soc., 2004,151(9):A1499-A1506
[2]Elumalai P,Vasan H N,Munichandraiah N.Mater.Res.Bull., 2004,39(12):1895-1907
[3]Tang H W,Zhu Z H,Chang Z R,et al.Electrochem.Solid-State Lett.,2008,11(3):A34-A37
[4]Ohzuku T,Makimura Y.Chem.Lett.,2001,30(8):744-745
[5]Ohzuku T,Makimura Y.Chem.Lett.,2001,30(7):642-643
[6]Kang S H,Kim J,Stoll M E,et al.J.Power Sources,2002, 112(1):414-48
[7]Kim J S,Johnson C S,Vaughey J T,et al.Chem.Mater, 2004,16(10):1996-2006
[8]Thackeray M M,Johnson C S,Vaughey J T,et al.J.Mater. Chem.,2005,15:2257-2267
[9]MacNeil D D,Lu Z,Dahn J R.J.Electrochem.Soc.,2002, 149(10):A1332-A1336
[10]Koyama Y,Tanaka I,Adachi H,et al.J.Power Sources, 2003,119-121(1):644-648
[11]Whitfield P S,Davidson I J,Cranswick L M D,et al.Solid State Lonics,2005,176(5/6):463-471
[12]ZHENG Jian-Ming(鄭建明),YANG Yong(楊勇).Proceedings of 14th Chinese Conference on Solid State Ionics Incorporating International Forum on Energy Storage&Conversion Technology(第十四屆全國固態離子學學術會議暨國際能量儲存與轉換技術論壇論文摘要集).Harbin:[s.n.], 2008:B38
[13]WU Xiao-Biao(吳曉彪),DONG Zhi-Xin(董志鑫),ZHENG Jian-Ming(鄭建明),et al.J.Xiamen Univ.:Nat.Sci.(Xiamen Daxue Xuebao),2008,47(Sup):224-227
[14]Zhang S,Deng C,Yang S Y,et al.J.Alloy Compd.,2009, 484(1/2):519-523
[15]Zhou F,Zhao X M,Bommel A,et al.Chem.Mater.,2010, 22(3):1015-1021
[16]Deng H X,Belharouak I,Cook R E,et al.J.Electrochem. Soc.,2010,157(4):A447-A452
[17]HU Wei(胡偉),XIE Hui(謝輝),ZHANG Qian(張騫),et al. World Nonfer.Metals(Shijie Youse Jinshu),2009,3:35-37
[18]Deng C,Zhang S,Ma L,et al.J.Alloy Compd.,2011,509 (4):1322-1327
[19]Yabuuchi N,Makimura Y,Ohzuku T.J.Electrochem.Soc., 2007,154(4):A314-A321
[20]Thackeray M M,Kang S H,Johnson C S,et al.Electrochem. Commun.,2006,8(9):1531-1538
[21]Wu Y,Manthiram A.Electrochem.Solid-State Lett.,2006,9 (5):A221-A224
Effects from Feeding Ways during Preparing Li[Li0.2Co0.13Ni0.13Mn0.54]O2by Carbonate Co-precipitation Method
DU Ke*ZHAO Jun-FengWANG Wei-GangHUANG Xia CAO Yan-Bing HU Guo-Rong PENG Zhong-Dong
(Institute of Light Metal and Industrial Electrochemistry,School of Metallurgical Science and Engineering,Central South University,Changsha410083,China)
The precursor of Mn0.675Ni0.1625Co0.1625CO3has been synthesized by a carbonate co-precipitation method, which was used to prepare a high capacity cathode material Li[Li0.2Mn0.54Ni0.13Co0.13]O2for lithium ion batteries. Three kinds of raw materials feeding ways during co-precipitation process were compared.The chemical and physical properties of the precursor and Li[Li0.2Mn0.54Ni0.13Co0.13]O2have been systematically studied.
lithium ion batteries;Li[Li0.2Co0.13Ni0.13Mn0.54]O2;cathode material;co-precipitation
O614.111;O614.7+11
A
1001-4861(2012)01-0074-07
2011-05-31。收修改稿日期:2011-08-05。
國家自然科學基金(No.50604018);中南大學中央高校基本科研業務費(No.2010QZZD0101)資助項目。*
。E-mail:duke22@csu.edu.cn

- 無機化學學報的其它文章
- Sonochemical Synthesis of Different Morphological CaF2Microstructures
- Synthesis,Crystal Structure and Properties of a Manganese(Ⅱ)Complex with an Asymmetrical Substituted Triaryltriazole
- S2-控制劑對Ag納米產物的形貌及光學性能的影響
- Syntheses and Crystal Structures of Nickeand CopperComplexes with Schiff Base Ligand of 5-Chlorosalicylaldehyde
- Hydrothermal Synthesis,Crystal Structure and Catalytic Properties of a Polyoxovanadate Organicamine
- Tumor-Imaging Core-Shell Nano-Models for Catalase