西侖吉肽的合成制備
2012-04-09 09:15:06賴伊麗黃世龍劉會敏
化學工業與工程 2012年4期
孔 毅,黃 海,賴伊麗,黃世龍,劉會敏
(中國藥科大學生命科學與技術學院,江蘇 南京 210009)
西侖吉肽(cilengitide,EMD- 121974)是德國化學家Kessler設計的含有RGD序列的環肽c[RGDf(N-Me)V]。該肽基于體內αvβ3整合素配體,通過綜合運用環化、空間篩選和甲基化掃描而得,對αvβ3整合素具有高親和性和選擇性[1]。臨床試驗表明,該肽不僅能較好抑制黑色素瘤和膠質細胞瘤生長,而且與放射治療配合能提高其他腫瘤如頭頸鱗狀細胞瘤的治愈率[2],具有較好的市場前景和潛力。
除西侖吉肽發明者提供合成方法外[1],目前尚未見西侖吉肽合成工藝細節研究的報道。本研究嘗試對西侖吉肽合成方法進行優化,采用Fmoc固相合成與液相合成相結合的方法,以HBTU,PyBOP/HOBt為縮合劑,對合成方法特別是難以縮合的氨基酸的縮合方法進行了研究,對環化條件進行了探索。本方法合成效率高、原料易得、路線簡單易行、具有一定的工業化應用前景。
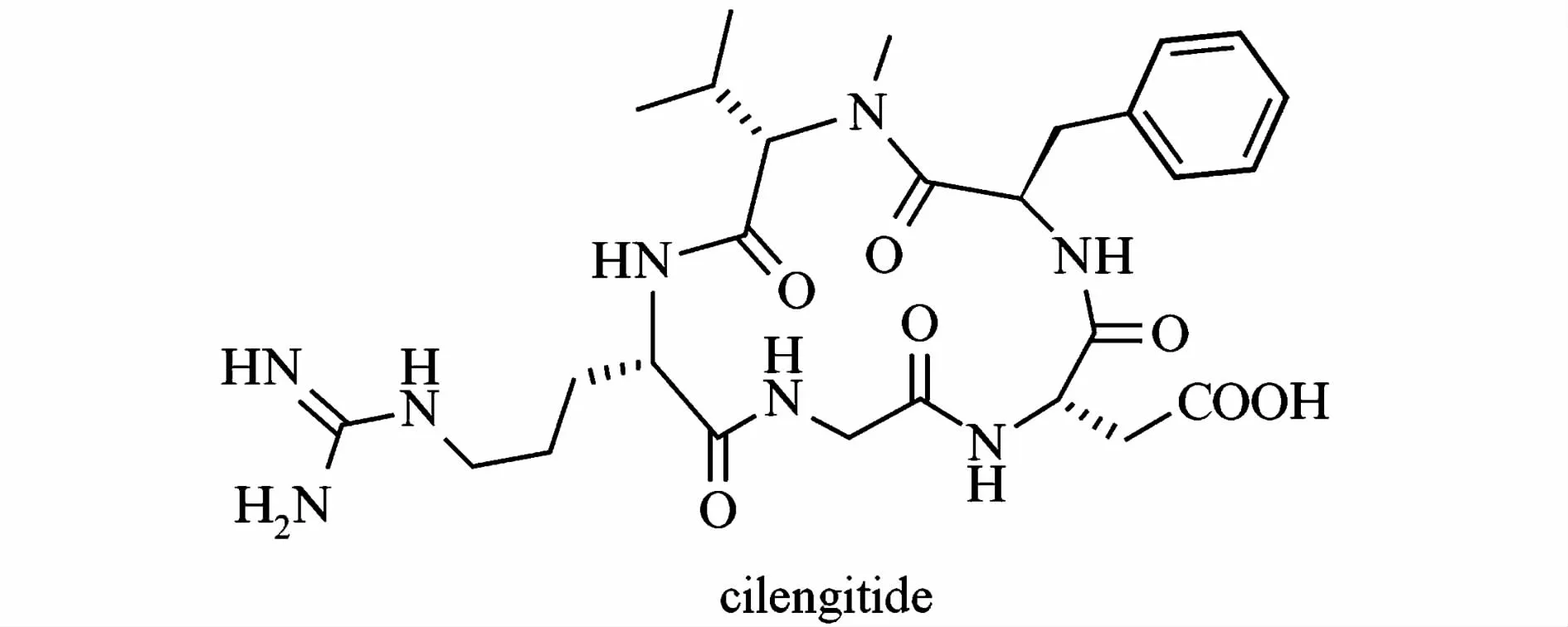
圖1 西侖吉肽分子結構式[1]Fig.1 The molecular structure of cilengitide[1]
1 材料與方法
1.1 儀器與設備
砂芯玻璃管,4號砂芯;循環水式真空泵,予華SHZ-DⅢ,河南鞏義予華儀器有限公司;紫外-可見分光光度計,752,上海菁華科技儀器有限公司;半制備型高效液相色譜儀,BioLogic DuoFlow Pathfinder 20/80 System,美國Bio-RAD;高效液相色譜儀,島津20A;凍干機,Labconco;質譜儀,島津2010MS。
1.2 材料與試劑
2-CTC樹脂,載量為0.9 mmol/g;9-芴甲氧基羰基(Fmoc)-氨基酸[Fmoc-Arg(Mtr)-OH;Fmoc-Gly-OH;Fmoc-(Otbu-)Asp-OH;Fmoc-D-Phe-OH;Fmoc-(N-Me-)Val-OH];苯并三氮唑-N,N,N’,N’-四甲基脲六氟磷酸酯(HBTU);六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷(PyBOP);1-羥基苯丙三氮唑(HOBT);N, N-二異丙基乙胺(DIEA) ;乙二硫醇(EDT);三異丙基硅烷(TIS),均為上海吉爾生化公司產品。二氯甲烷(DCM);無水甲醇;無水乙醇;無水乙醚;三氟乙酸(TFA);六氫吡啶(哌啶,PIP);N, N-二甲基甲酰胺(DMF);茚三酮;均為國產分析純。乙腈,美國TEDIA,色譜純;純凈水,樂百氏牌。
1.3 方法
1.3.1樹脂溶脹
稱取2 g 2-CTC樹脂于燒杯中,用20 mL DMF浸泡30 min后倒出約一半體積DMF,加入10 mL DCM浸泡30 min,用DMF洗滌樹脂2~3次后,用真空泵將樹脂抽濾干。
1.3.2線性肽的合成
1)首個氨基酸上載:稱取Fmoc-Arg(Mtr)-OH 4.16 g溶于10 mLDMF,再加入DIEA 2 mL,溶解完全后與樹脂混合均勻,通氮氣室溫25 ℃下攪拌反應2 h;取少量于EP管中,加20%(體積比)PIP/DMF脫保護5 min,茚三酮法(Kaiser法)[3]定性檢測反應進行的程度,判斷各步接肽的終點,反應結束后,加入2 mL甲醇,繼續反應0.5 h后,抽濾掉反應液后。以DMF洗滌2次后,再以DMF,無水甲醇交替洗滌2次。真空干燥稱重測肽樹脂擔載率,mmol/g。
2)脫保護:加入30 mL 20%PIP/DMF浸沒肽樹脂,氮氣攪拌,分別經過5和15 min的2次脫保護后,茚三酮法檢測,反應完全后抽濾掉反應液,DMF洗滌樹脂3~4次,抽干。
3)上載下一個氨基酸:稱取Fmoc-(N-Me-)Val-OH 1.20 g 溶于適量DMF,再加入HBTU 1.29 g,HOBt 0.5 g,DIEA 1 mL溶解完全后與肽樹脂混合均勻,通氮氣室溫25 ℃下攪拌反應2 h;茚三酮法檢測,反應完全后抽濾掉反應液,DMF洗滌3~4次。Fmoc-(N-Me-)Val-OH為困難氨基酸,單純使用HBTU不能完全縮合,嘗試改用縮合劑PyBOP和延長反應時間來完成縮合。
4)上載其他3個氨基酸:重復2)和3),把剩下的Fmoc-D-Phe-OH、Fmoc-(Otbu-)Asp-OH和Fmoc-Gly-OH依次接到肽樹脂上。
1.3.3Fmoc法[4]測定連接效率
稱取Fmoc-Gly-OH(相對分子質量:339.4)5 mg置于小玻璃瓶,加1 mL體積分數為20%的PIP/DMF,脫保護30 min,無水甲醇定容至100 mL;再稀釋10倍后, 依次取1、2、3、5、6 和 7 mL置于10 mL容量瓶中,無水甲醇定容,分別與空白即體積分數為20%的PIP/DMF對照,測301 nm的紫外吸收值。利用標準曲線計算肽樹脂的縮合效率。
1.3.4肽樹脂切割與肽沉淀
在樹脂上將線性肽合成完成后,用DMF洗滌3次,甲醇洗滌1次,DMF 洗滌1次,甲醇洗滌1次后真空干燥后,稱重,按1 g肽樹脂5 mL裂解液TFA/DMF(體積比5∶95)的比例加入裂解液,冰浴攪拌反應2 h。然后將裂解液抽濾,并用少量裂解液沖洗樹脂2~3次,沖洗液和裂解液收集到一起。加無水乙醚沉淀得到線性肽粗品,HPLC/MS分析。
1.3.5環肽的合成
線性肽粗品0.85 g溶解于2 000 mL DCM中,加入PyBOP 1.52 g,HOBt 0.4 g,和DIEA 1.2 mL,冰浴攪拌反應24 h。將反應液減壓旋蒸至約20 mL。分別用10%HCl/H2O(體積比),飽和NaHCO3,飽和NaCl依次萃取后,加入等體積裂解液TFA∶TIS∶EDT∶H2O為95∶2∶2∶1冰浴反應2 h。無水乙醚沉淀得環肽粗品。
1.3.6環肽的分離純化和鑒定
用高效液相色譜(HPLC)分析環肽粗品,液相色譜條件: Kromasil C18(4.6 mm×250 mm) 色譜柱; 波長λ為214 nm; 流動相,A為純凈水∶乙腈∶TFA為90.0∶10.0∶0.1 (體積比), B為乙腈∶純凈水∶TFA為90.0∶10.0∶0.1 (體積比);進樣量10 μL; 流速1 mL/min。經MS分析確定主峰與目標肽相對分子質量一致后,用制備型高效液相色譜(HPLC)純化環肽粗品后,冷凍干燥。純化后的環肽用分析HPLC檢測純度,并用質譜與核磁共振研究其結構。
2 結果與分析
2.1 樹脂的選擇
選擇2-CTC樹脂(分子式見圖2, P表示樹脂珠),因為一方面,與其他樹脂相比,2-CTC樹脂具有很強的活性;氨基酸羧基端與2-CTC樹脂的反應為取代反應,是不可逆反應。另一方面,由于2-CTC樹脂反應位點附近位阻較大,大大減少了二肽時由于分子內環化生成哌嗪二酮(DKP)[5]的可能性。
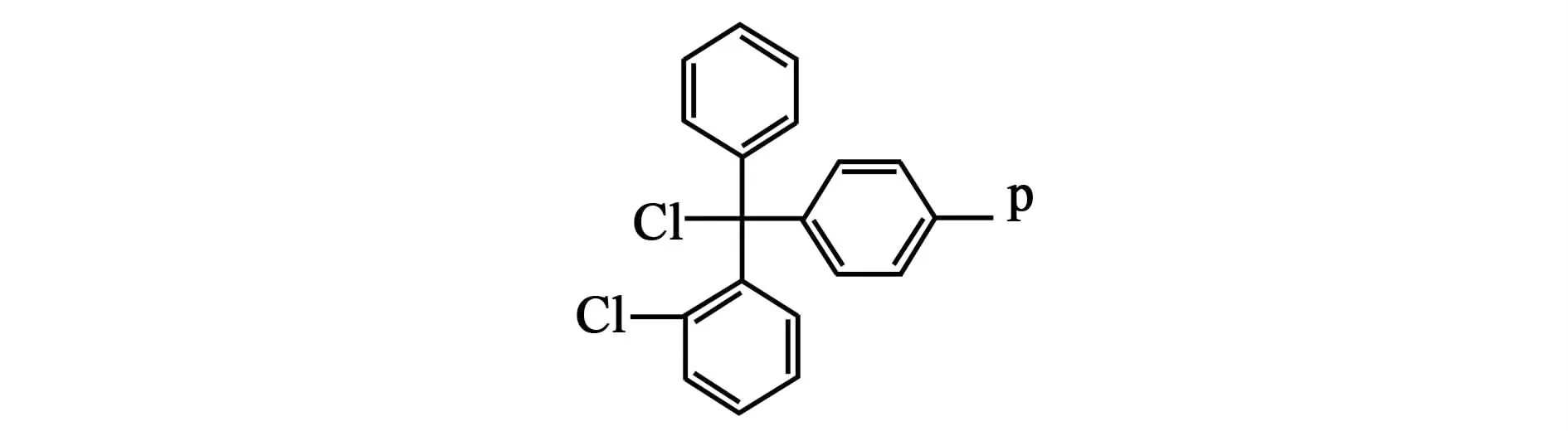
圖2 二氯樹脂Fig.2 2-CTC resin
2.2 接肽反應
普通氨基酸用HBTU的縮合效率達到95%以上;N-甲基氨基酸屬于困難氨基酸,其反應活性很低,為提高共縮合效率,分別考察HBTU和PyBOP在不同時間2,4,6,8,10 h的縮合效率,結果如圖3。
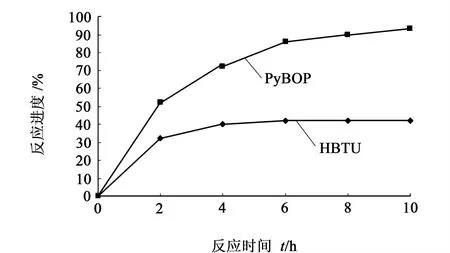
圖3 HBTU和PyBOP反應進度圖Fig.3 The extent of reaction with HBTU & PyBOP
確定8 h為最適宜反應時間后,考慮到HBTU有一定的縮合效率,而PyBOP雖然縮合效率高但價格較貴,于是采用HBTU和PyBOP以一定比例混合使用,分別以HBTU∶PyBOP(體積比)為1∶2,1∶1,2∶1,3∶1考察縮合效率,試驗結果顯示2∶1為最適宜配比。
2.3 α-氨基保護集基團的脫除
在每一步接肽反應完成后, 下一個氨基酸接入之前, 需要除去保護基團。本次試驗采用的是PIP/DMF的溶液,基團的脫除效果與哌啶的含量及其脫除時間關系見圖4。

圖4 不同濃度哌啶Fmoc脫除率Fig.4 The Fmoc deprotection ratio of piperidine at different concentration
圖4可以看出,隨著哌啶濃度的增加,脫保護率增加且脫保護的時間縮短。使用20%濃度的保護試劑在10 min時脫保護效率可達99%。
2.4 線性肽粗品HPLC和MS結果分析
線性肽粗品HPLC和MS分析結果見圖5和圖6。色譜條件:流動相A為乙腈/水(10%,含0.1 %TFA ,體積分數,下同),B為乙腈/水(90%,含0.1% TFA),梯度洗脫0~30 min,20%~40% B。紫外檢測器,波長范圍為214 nm;色譜柱為Kromasil C18,規格為4.6×250 mm,流速為0.2 mL/min。樣品進樣量為10 μL 。
質譜條件:離子檢測方式SCAN,離子極性正離子,離子化方式為電噴霧離子化,檢測電壓1.5 kV,霧化氣體流速1.5 L/min;曲線脫溶劑裝置(CDL)溫度250 ℃,加熱塊(block)溫度200 ℃。

圖5 線性肽粗品HPLC圖Fig.5 The HPLC of the crude linear cilengitide
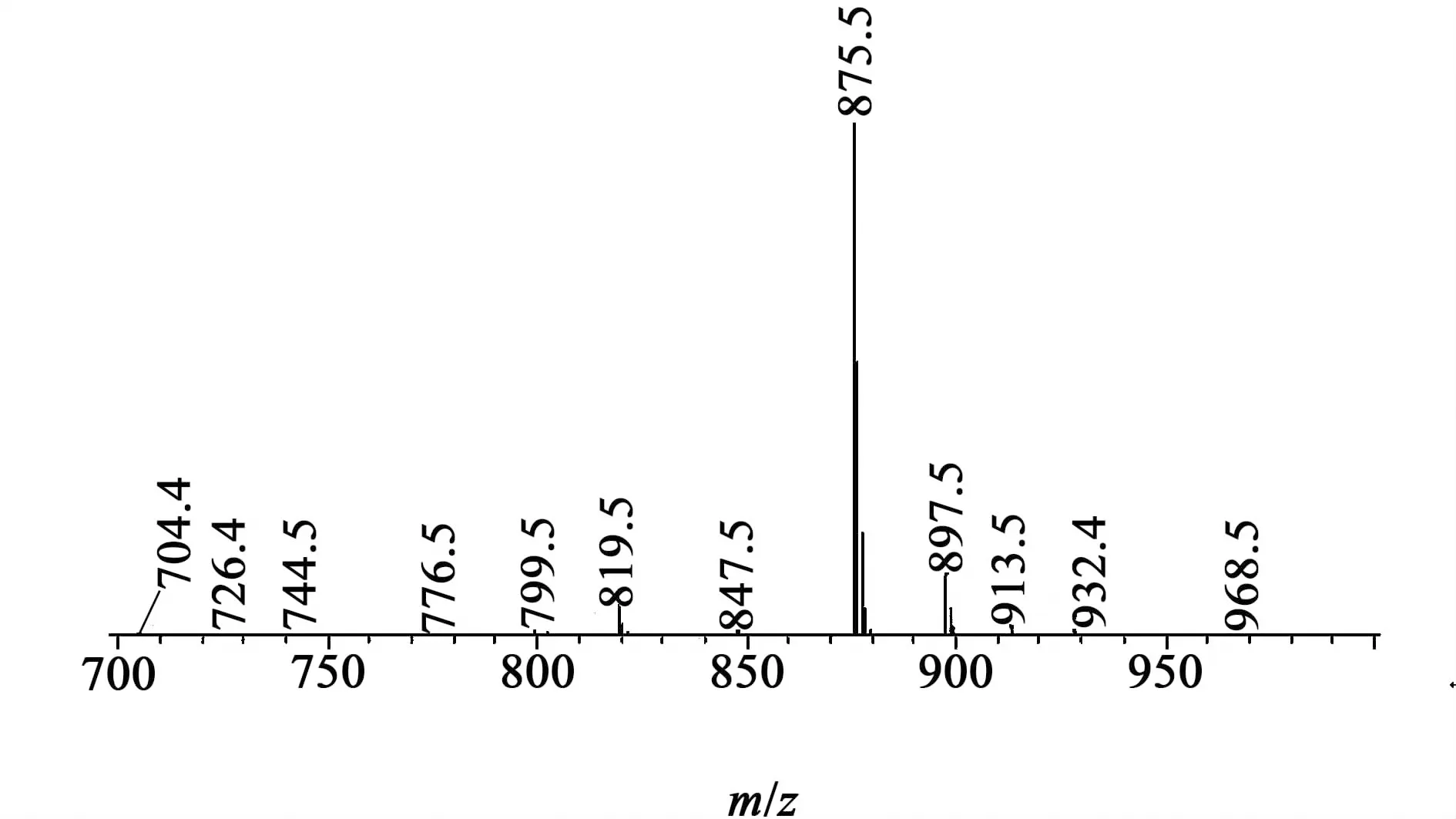
圖6 線性肽粗品主峰質譜圖Fig.6 The MS of the crude linear cilengitide
2.5 環化方法的選擇
一般環肽按照成環所含鍵分為2類:全部為酰胺鍵的均環肽為一類,除酰胺鍵外還有二硫鍵和醚鍵等的雜環肽為另一類。而按照橋連位置分的話,分為頭尾相連環肽、頭側相連環肽、側側相連環肽。其中頭尾相連合成難度最大,因為線性肽分子趨于能量較低的穩定形態,也就是舒展的狀態。這樣分子的反應位點,即兩端的羧基和氨基空間相距較遠,靠近的可能性較低,反應活性降低。
液相合成環肽,其中發生的反應主要是分子內的反應和分子間的反應。為了減少副反應分子間反應形成的多聚體,應該盡量將肽反應濃度降低,一般控制在10-3~10-4mol/L高度稀釋的溶液里反應。常用的液相合成法有:活潑酯法、疊氮法、混合酸酐法、直接法、硫酯法和輔助成環法[6]。其中活潑酯法、疊氮法和混合酸酐法需要制備中間體,且需要低溫反應。根據實際情況,最終確定使用直接法,反應濃度控制范圍1×10-3~1×10-4mol/L,環化過程用HPLC檢測至反初始應物峰消失。
2.6 環肽粗品的HPLC分析結果
沉淀后得白色環肽粗品560 mg,HPLC分析結果如圖7。
色譜條件:流動相A為乙腈/水(10%,含0.1%TFA,體積分數,下同),B為乙腈/水(90%,含0.1% TFA) ,梯度洗脫為0~30 min,10%~100% B,紫外檢測器,波長范圍為214 nm;色譜柱為反相C18,規格為4.6×250 mm,流速為0.2 mL/min。樣品進樣量為10 μL 。
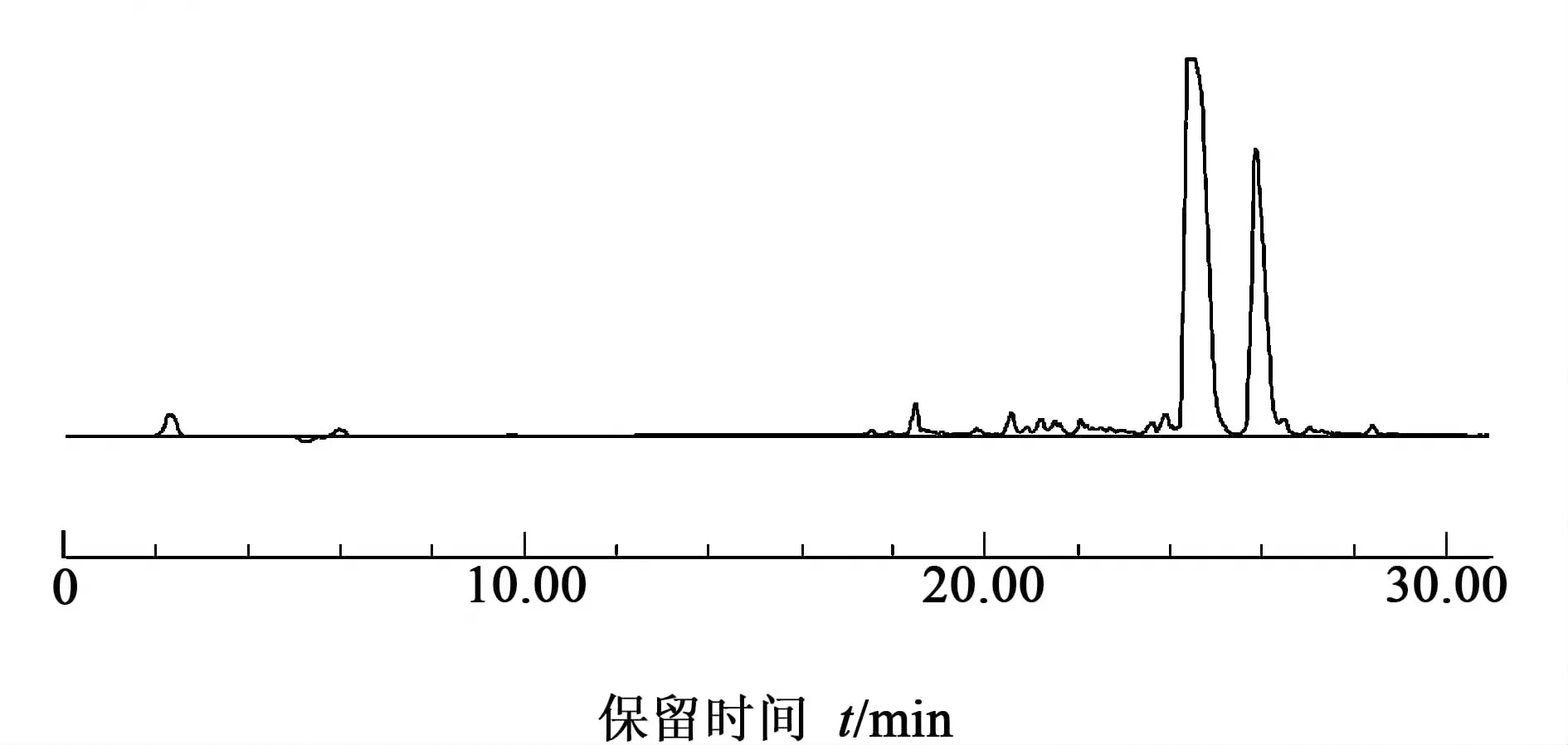
圖7 環肽粗品HPLC圖Fig.7 The HPLC of the crude cilengitide
2.7 環肽純化后的HPLC、MS和NMR分析結果
目標峰液體真空冷凍干燥得到189 mg 西侖吉肽,在粗品中質量分數為33.8%;西侖吉肽純度為98.3%,總產率為28.2%(以首個氨基酸上載量1.14 mmol記)。
HPLC結果見圖8,MS結果見圖9。 NMR結果與文獻報道的NMR圖譜[11]一致。

圖8 環肽純化后HPLC圖Fig.8 The HPLC of the purified cilengitide

圖9 環肽純化后質譜圖Fig.9 The MS of the purified cilengitide
3 結論
本研究采用Fmoc固相法合成線性肽,然后采用液相法環化合成了西侖吉肽。合成中,解決了難以縮合氨基酸的縮合效率問題,合成了高純度的線性肽;并選擇合適的液相環化條件合成環肽。所合成環肽純度為98.3%,相對分子質量與理論相對分子質量588相符,NMR圖譜與文獻報道一致,說明該方法合成的西侖吉肽結構正確。本方法合成效率高,原料易得,路線簡單易行,具有一定的工業化應用前景。
參考文獻:
[1]MAS-MORUNO C,RECHENMACHER F,KESSLER H.Cilengitide:The first anti-angiogenic small molecule drug candidate.Design,synthesis and clinical evaluation[J].Anti-Cancer Agents in Medicinal Chemistry,2010,10:753-768
[2]HEHLGANS S,MICHAEL H,NILS C.Signalling via integrins:Implications for cell survival and anticancer strategies[J].Biochimica et Biophysica Acta,2007,(1775):163-180
[3]KALSER E,COLESCOTT R L,BOSSING C D,etal.Color test for detection of terminal amino groups in the solid phase synthesis of peptides[J ].Anal Biochem,1970,34:595-598
[4]張俊,楊明,王安明,等.微波作用下大位阻氨基酸與H-Pro-CTC 樹脂的高效縮合[J].有機化學,2008,28,(12): 2 119-2 125
[5]林浩,王德心.哌嗪二酮衍生物的合成研究進展[J].藥學學報,2003,38(5):395-400
[6]唐艷春,田桂玲,葉蘊華.環肽合成方法的研究進展[J].高等學校化學學報,2000,21(7):1 056-1 063
[7]吳蕾,任云霞,景文鵬.神經降壓素的固相合成工藝研究[J].化學工業與工程,2009,26(4):292-297
[8]黃惟德,陳常慶.多肽合成[M].北京:科學出版社,1985
[9]王德心.固相有機合成:原理及應用指南[M].北京:化學工業出版社,2004
[10]SEWALD N,JAKUBKE H J.Peptides-Chemistry and biology[M].Germany:Wiley-VCH,2002
[11]CUPIDO T,SPENGLER J,RUIZ-RODRIGUEZ J,etal.Amide-to-ester substitution allows fine-tuning of the cyclopeptide conformational ensemble[J].Angew Chem Int Ed Engl,2010,49(15):2 732-2 737
[12]CLARK R J,AKCAN M,KAAS Q,etal.Cyclization of conotoxins to improve their biopharmaceutical properties[J/OL].Toxicon (2010),doi:10.1016/j.toxicon.2010-12-03
[13]DAI X,SU Z,LIU J.An improved synthesis of a selectiveαvβ3-integrin antagonist cyclo(-RGDfK-)[J].Tetrahedron Letters,2000(41):6 295-6 298
