馬方綜合征合并左室心肌致密化不全1例報道
2012-01-26 08:00:20杜志峰侯瑞田丁華杰王勝林承德醫學院附屬醫院骨傷科心內科超聲科影像科河北承德067000
重慶醫學 2012年18期
杜志峰,侯瑞田,丁華杰,王勝林(承德醫學院附屬醫院:.骨傷科;.心內科;.超聲科;.影像科,河北承德 067000)
馬方綜合征合并左室心肌致密化不全1例報道
杜志峰1,侯瑞田2,丁華杰3,王勝林4
(承德醫學院附屬醫院:1.骨傷科;2.心內科;3.超聲科;4.影像科,河北承德 067000)
1 臨床資料
患者,女,15歲,因父母發現其持物不穩及不能完成跳繩動作而就診,追問患者偶發頭痛?查體:體形消瘦,眼球前凸,頸靜脈無怒張,兩側胸廓不對稱,漏斗胸(輕度),雙肺呼吸音清,未聞及干濕性啰音;心尖搏動范圍正常,心率80次/分,心臟相對濁音界正常,各瓣膜聽診區未聞及雜音?周圍血管無毛細血管搏動及水沖脈?腹部及神經檢查陰性,雙下肢無水腫?蜘蛛指(雙手纖長),指征及腕征陽性,脊柱側彎,足弓過高,關節松弛,雙上肢肌力正常,右下肢肌力4級,左下肢肌力5級,各病理征陰性?血?尿常規及各項生化檢查未見異常;24h動態心電圖示偶發房性期前收縮,見圖1;心臟彩超示左心室心肌致密化不全,見圖2;肺部平片未見異常,脊柱正?側位X線片示脊椎側彎,見圖3;脊髓MRI示頸2至胸1硬膜囊擴張,見圖4?診斷:馬方綜合征,左心室心肌致密化不全?

圖1 24h動態心電圖示偶發房性期前收縮
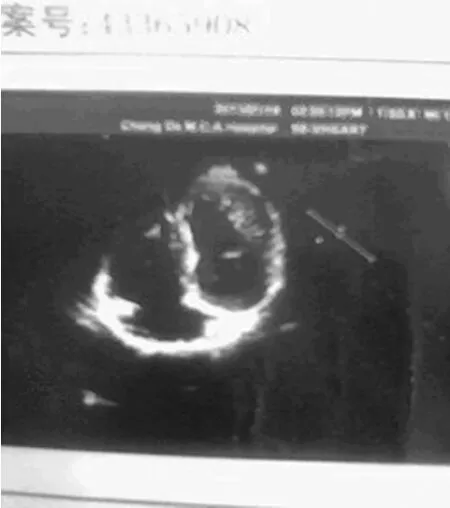
圖2 心臟彩超示左心室心肌致密化不全
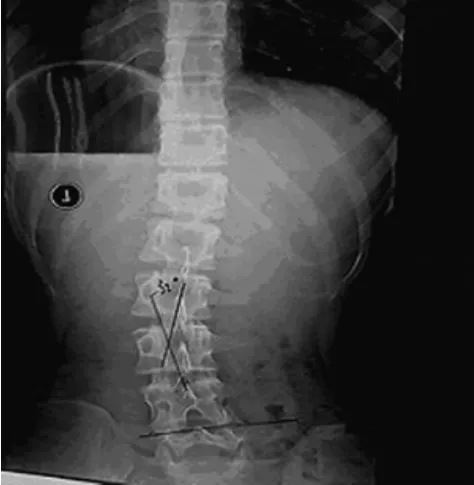
圖3 脊柱正位X線片示腰椎側彎
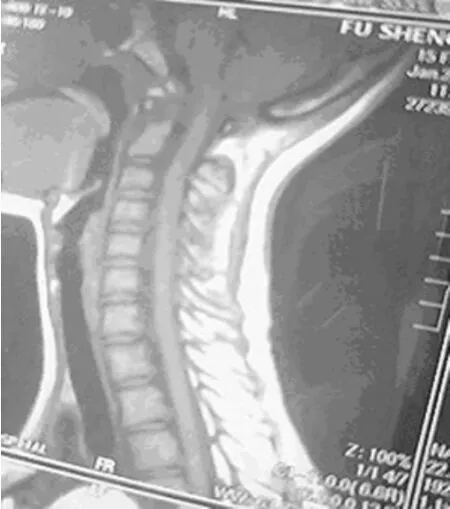
圖4 頸髓MRI示硬膜囊輕度擴張
2 討 論
馬方綜合征是人類第15號常染色體長臂上FBN1基因缺陷所致的全身性結締組織遺傳病[1]?臨床主要以心血管?眼和骨骼肌肉系統受累為主,其中30.00%~60.00%的患者有血管系統異常?馬方綜合征患者心血管系統病變中主動脈根部擴張占93.80%,主動脈瓣關閉不全占87.50%,主動脈夾層占46.90%,二尖瓣脫垂占12.50%,主動脈竇瘤占6.20%,肺動脈擴張占3.10%,心包積液占12.50%,二葉主動脈瓣占6.20%,房間隔缺損占3.10%,主動脈縮窄占1.00%[2]?
心肌致密化不全是一種罕見的原發性心肌病?2006年美國心臟病協會(AHA)將心肌致密化不全歸為遺傳性心肌病,2008年歐洲心臟病協會(ESC)將其歸為未定型心肌病?心肌致密化不全的發病率為0.05%~0.24%[3],有家族聚集傾向,日本家族發病率為44.00%,中國為18.00%~40.00%[4-5]?有研究認為心肌致密化不全與常染色體18q?11q?10q基因突變有關[6-8]?心肌致密化不全臨床癥狀出現的早晚及輕重與病變范圍有關,以心力衰竭?心律失常?血栓栓塞為三大臨床表現,患者心臟性猝死發生率高達13.00%~18.00%[9-11],室性快速性心動過速被認為是一種最常見的致死原因,大多數心肌致密化不全患者在確診時已具有室性心律失常及心力衰竭癥狀,而本病多與生俱來,故而早期診斷將直接影響患者的生存率,早期診斷并早期應用植入性心臟除顫器(ICD)對患者(尤其是有室性快速性心律失常患者)的預后均大有裨益[12-14]?以往研究未發現心肌致密化不全患者合并有典型馬方綜合征[15],而眾多關于馬方綜合征心臟病報道中,亦無心肌致密化不全記載[2],本例的發現無疑對二者互相參診具有重要意義?
[1] Giampietro PF,Raggio C,Davis JG.Marfan syndrome:orthopedic and genetic review[J].Curr Opin Pediatr,2002,14(1):35-41.
[2] 宋芝萍,劉旭,倪幼芳.馬方綜合征心血管系統臨床表現的研究[J].中國實用內科雜志,2000,20(9):542-543.
[3] St?llberger C,Finsterer J,Blazek G.Left ventricular hypertrabeculation/noncompaction and association with additional cardiac abnormalities and neuromuscular disorders[J].Am J Cardiol,2002,90(8):899-902.
[4]Ichida F,Hamamichi Y,Miyawaki T,et al.Clinical features of isolated noncompaction of the ventricular myocardium:long-term clinical course,hemodynamic properties,and genetic background[J].J Am Coll Cardiol,1999,34(1):233-240.
[5] 夏利,李華,劉良東.心肌致密化不全一家系[J].中華醫學遺傳學雜志,2004,21(3):301-301.
[6] Bowles KR,Gajarski R,Porter P,et al.Gene mapping of familial autosomal dominant dilated cardiomyopathy to chromosome 10q21-23[J].J Clin Invest,1996,98(6):1355-1360.
[7]Ichida F,Tsubata S,Bowles KR,et al.Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome[J].Circulation,2001,103:1256-1263
[8] Sasse-Klaassen S,Probst S,Gerull B,et al.Novel gene locus for autosomal dominant left ventricular noncompaction maps to chromosome 11p15[J].Circulation,2004,109(22):2720-2723.
[9] Chin TK,Perloff JK,Williams RG,et al.Isolated noncompaction of left ventricular myocardium.A study of eight cases[J].Circulation,1990,82(2):507-513.
[10]Zambrano E,Marsh AS,Jaffe CC,et al.Iso lated non compact ion of the ventricularmyocardium:Cl inical and molecular aspects of a rare cardiomyopathy[J].Lab Invest,2002,82(2):117-122.
[11]Oechslin EN,ttenhofer-Jost AT,Rojas JR,et al.Longterm follow up of 34adults with isolated left ventricular non compaction:A distinct cardiomyopathy with poor prognosis[J].J Am Coll Cardiol,2000,36(2):493-500.
[12]Chan YC,Ting CW,Ho P,et al.Ten-year epidemiological review of in-hospital patients with Marfan syndrome[J].Ann Vasc Surg,2008,22(5):608-612.
[13]Kobza R,Jenni R,Erne P,et al.Implantable cardioverter-defibrillators in patients with left ventricular noncompaction[J].Pacing Clin Electrophysiol,2008,31(4):461-467.
[14]Murphy RT,Thaman R,Blanes JG,et al.Natural history and familial characteristics of isolated left ventricular noncompaction[J].Eur Heart J,2005,26(2):187-192.
[15]Tsai SF,Ebenroth ES,Hurwitz RA,et al.Is left ventricular noncompaction in children truly an isolated lesion?[J].Pediatr Cardiol,2009,30(5):597-602.
10.3969/j.issn.1671-8348.2012.18.048
C
1671-8348(2012)18-1892-03
2011-08-24
2011-12-22)
?臨床護理?
