一種酵母快速批量分子鑒定方法*
2011-12-18 11:23:06陳源源石貴陽(yáng)王正祥
食品與發(fā)酵工業(yè) 2011年2期
陳源源,石貴陽(yáng),王正祥
1(江南大學(xué)生物工程學(xué)院工業(yè)生物技術(shù)教育部重點(diǎn)實(shí)驗(yàn)室,江蘇無(wú)錫,214122)2(江南大學(xué)中國(guó)高校工業(yè)微生物資源與信息中心,江蘇無(wú)錫,214122)
一種酵母快速批量分子鑒定方法*
陳源源1,2,石貴陽(yáng)1,2,王正祥1,2
1(江南大學(xué)生物工程學(xué)院工業(yè)生物技術(shù)教育部重點(diǎn)實(shí)驗(yàn)室,江蘇無(wú)錫,214122)2(江南大學(xué)中國(guó)高校工業(yè)微生物資源與信息中心,江蘇無(wú)錫,214122)
2 6S rDNA D1/D2區(qū)域序列同源性分析是一種常用的酵母分子鑒定方法。D1/D2區(qū)域序列的擴(kuò)增需要酵母染色體DNA作為模板,目前常用的酵母染色體DNA提取方法繁瑣耗時(shí)且難以進(jìn)行批量操作,限制了酵母分子鑒定的規(guī)模化。研究中建立了一種快速高效的批量酵母染色體DNA提取方法,整個(gè)提取過(guò)程僅耗時(shí)20 min,從而使得酵母大規(guī)模快速分子鑒定成為可能。用該方法制備的染色體DNA不需要任何后續(xù)處理即可作為模板用于擴(kuò)增酵母的26S rDNA D1/D2區(qū)域序列,獲得了良好的擴(kuò)增結(jié)果以及擴(kuò)增產(chǎn)物的測(cè)序結(jié)果。
酵 母,染色體DNA,提取,26S rDNA D1/D2區(qū)域,鑒定
酵母是一類(lèi)單細(xì)胞真菌的統(tǒng)稱(chēng),目前已知的1 000多種酵母,大部分被歸類(lèi)到子囊菌門(mén)。作為重要的微生物資源,酵母是微生物多樣性的重要組成部分,收集和整理酵母菌株對(duì)保護(hù)遺傳資源和生物多樣性具有重要意義。對(duì)酵母進(jìn)行分類(lèi)鑒定是實(shí)現(xiàn)其資源化的基礎(chǔ),傳統(tǒng)的酵母鑒定方法包括形態(tài)學(xué)鑒定和生理生化鑒定[1]。由于酵母一般以單細(xì)胞形態(tài)存在,能用于種屬鑒定的形態(tài)特征很少,因此僅依靠形態(tài)觀(guān)察難以對(duì)酵母進(jìn)行有效鑒定。常用的酵母生理生化鑒定方法需要60~90項(xiàng)實(shí)驗(yàn),完成整個(gè)鑒定過(guò)程往往需要幾周的時(shí)間[2]。盡管酵母生理生化鑒定方法目前仍被廣泛使用,但一些種屬相近的酵母代謝途徑相似,且部分生理生化特征隨著環(huán)境的變化而不穩(wěn)定,因此僅僅通過(guò)生理生化實(shí)驗(yàn)對(duì)酵母進(jìn)行種屬鑒定是不夠準(zhǔn)確的[3]。
隨著聚合酶鏈?zhǔn)椒磻?yīng)技術(shù)(PCR)以及DNA測(cè)序技術(shù)的發(fā)明,分子生物學(xué)被越來(lái)越多的用于酵母的分類(lèi)與鑒定[4]。目前核糖體DNA(rDNA)序列分析法在微生物系統(tǒng)分類(lèi)中被廣泛應(yīng)用,通過(guò)26S rDNA的D1/D2區(qū)域序列同源性分析對(duì)酵母進(jìn)行分類(lèi)鑒定已經(jīng)被大多數(shù)研究者所接受和采用[5-7]。D1/D2區(qū)域序列的PCR擴(kuò)增需要酵母染色體DNA作為模板。目前常用的酵母染色體DNA提取方法包括煮沸法[8]、石英砂研磨法[9]、SDS 裂解法[10]和酶解法[11]等,其中煮沸法雖然耗時(shí)較短,但其提取成功率較低,且不適用于提取某些種屬酵母的染色體DNA;而其它幾種提取方法的步驟復(fù)雜,需要耗費(fèi)較長(zhǎng)的時(shí)間,且難以進(jìn)行批量操作,這些都成為制約酵母分子鑒定規(guī)模化的瓶頸。
本文建立了一種新的快速批量提取酵母染色體DNA的方法,整個(gè)提取過(guò)程僅耗時(shí)20 min。用該方法制備的染色體DNA無(wú)需任何純化處理即可作為PCR模板,用于擴(kuò)增酵母26S rDNA的D1/D2區(qū)域序列。
1 材料與方法
1.1 實(shí)驗(yàn)儀器
Alpha EC凝膠成像系統(tǒng),美國(guó)Alpha innotech公司;PTC-200 PCR儀,美國(guó)Bio-Red公司;HB100恒溫金屬浴,杭州博日科技有限公司;1~15臺(tái)式高速離心機(jī),德國(guó)Sigma公司;Vortex Genius 3振蕩器,德國(guó)IKA公司;水平電泳儀,北京六一廠(chǎng);3130基因測(cè)序儀,美國(guó)Applied Biosystems公司。
1.2 生化試劑
Ex Taq DNA聚合酶、dNTP Mixture(2.5mol/L)和DL2000 DNA marker購(gòu)自寶生物有限公司;PCR產(chǎn)物純化試劑盒購(gòu)自美國(guó)Axygen公司;BigDye Terminator v3.1 cycle sequencing kit試劑盒為美國(guó)Applied Biosystems公司產(chǎn)品;酵母裂解液由江南大學(xué)中國(guó)高校工業(yè)微生物資源與信息中心配制。
1.3 供試菌種
研究中使用的17株酵母菌株見(jiàn)表1,供試菌株由江南大學(xué)中國(guó)高校工業(yè)微生物資源和信息中心(http://cicim-cu.jiangnan.edu.cn)提供。
1.4 培養(yǎng)基及培養(yǎng)條件
將酵母菌株從甘油管中接出,在YPD培養(yǎng)基平板上劃線(xiàn),30℃培養(yǎng)。

表1 供試菌株名稱(chēng)
1.5 酵母染色體DNA的快速制備
在1.5 mL離心管中加入200 μL無(wú)菌水,從平板上刮取3~4環(huán)新鮮的酵母菌體于離心管中。振蕩洗滌后8 000×g離心1 min,吸盡上清液。在離心管中加入酵母裂解液100 μL,振蕩混勻后置于恒溫金屬浴中85℃反應(yīng)15 min,得到酵母染色體DNA粗提液。
1.6 26S rDNA的D1/D2區(qū)域的擴(kuò)增及PCR產(chǎn)物純化
在PCR薄壁管中加入上述酵母染色體DNA粗提液1 μL,10 × PCR 緩沖液 5 μL、dNTP Mixture 4 μL、30 μmol/L 的26S rDNA D1/D2 區(qū)域序列上下游通用引物(pNL1:5'-GCATATCAATAAGCGGAGGAAAAG-3 ';pNL4:5 '-GGTCCGTGTTTCAAGACGG-3 ')各 0.5 μL[12]、Ex Taq DNA 聚合酶 0.2 μL,補(bǔ)無(wú)菌水至50 μL。PCR反應(yīng)條件為:94℃預(yù)變性5 min;94℃變性30 s,53℃退火45 s,72℃延伸1 min,經(jīng)過(guò)30個(gè)循環(huán)后,72℃再延伸6 min。
取5 μL PCR產(chǎn)物在1﹪的瓊脂糖凝膠上進(jìn)行電泳檢測(cè),電泳結(jié)束后用凝膠成像系統(tǒng)觀(guān)察結(jié)果。PCR產(chǎn)物純化參照Axygen公司PCR產(chǎn)物純化試劑盒說(shuō)明書(shū)進(jìn)行。
1.7 核苷酸序列測(cè)定及比對(duì)
核苷酸序列測(cè)定參照BigDye Terminator v3.1試劑盒說(shuō)明書(shū)進(jìn)行。將測(cè)序結(jié)果用Sequence Scanner V1.0軟件進(jìn)行分析,查看序列的長(zhǎng)度及可信度。選擇測(cè)序結(jié)果中可信度較高的區(qū)域,提交至NCBIBLAST網(wǎng)站進(jìn)行比對(duì)。
2 結(jié)果與分析
2.1 酵母染色體DNA快速提取方法的建立
本研究建立了一種新的酵母染色體DNA快速批量提取方法,制備的染色體DNA無(wú)需純化處理即可直接用于后續(xù)的PCR反應(yīng)。在研究過(guò)程中對(duì)染色體DNA快速提取方法中酵母裂解液的配制及其廣泛適用性進(jìn)行了反復(fù)研究和分析。裂解液由2 mol/L的NaOH溶液組成,并用非離子型濃度相關(guān)性pH緩沖劑小心調(diào)節(jié)其pH值至13.4左右。
2.2 提取的酵母染色體DNA質(zhì)量分析
2.2.1 PCR產(chǎn)物質(zhì)量檢驗(yàn)
取PCR產(chǎn)物5 μL進(jìn)行瓊脂糖凝膠電泳,17個(gè)樣品在約600 bp處均出現(xiàn)單一的目的條帶(圖1)。證明成功擴(kuò)增了供試酵母菌株的26S rDNA的D1/D2區(qū)域序列。

圖1 供試酵母菌株26S rDNA的D1/D2區(qū)域序列擴(kuò)增產(chǎn)物電泳圖
2.2.2 不同模板加樣量對(duì)PCR反應(yīng)的影響
在50 μL的PCR反應(yīng)體系中,分別加入0.25、0.5、1、1.5、2 和2.5 μL 的酵母染色體DNA 粗提液作為模板進(jìn)行PCR反應(yīng)。電泳后發(fā)現(xiàn)不同模板加樣量的PCR產(chǎn)物在約600 bp處均有明亮的單一條帶,條帶亮度基本相同,無(wú)非特異性條帶出現(xiàn),依次對(duì)應(yīng)于圖2中的1~6號(hào)泳道。
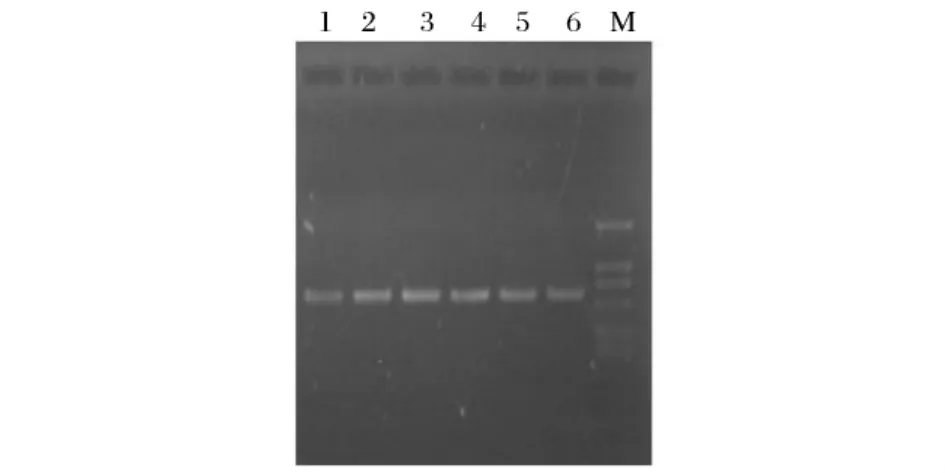
圖2 不同模板加樣量的PCR產(chǎn)物電泳圖
2.2.3 制備的染色體DNA的穩(wěn)定性分析
使用在4℃放置2周的酵母染色體DNA粗提液作為PCR模板擴(kuò)增ITS序列,電泳后在約600 bp處出現(xiàn)明亮單一的條帶。
2.3 PCR產(chǎn)物核苷酸序列測(cè)定及比對(duì)結(jié)果
上述獲得的PCR產(chǎn)物經(jīng)純化后,用于核苷酸序列測(cè)定。將測(cè)序結(jié)果用Sequence Scanner V1.0軟件分析后發(fā)現(xiàn)測(cè)得的信號(hào)清晰,無(wú)雜峰出現(xiàn)。
將測(cè)序結(jié)果提交至NCBI-BLAST網(wǎng)站進(jìn)行序列同源性比對(duì),發(fā)現(xiàn)17株酵母菌株的比對(duì)結(jié)果與原始種屬一致。
3 討論
酵母染色體DNA提取的實(shí)質(zhì)是破壞其細(xì)胞壁,釋放出染色體DNA。傳統(tǒng)的酵母染色體DNA提取首先要液體搖瓶培養(yǎng)酵母菌體,離心收集菌體后再用物理或化學(xué)方法破壞酵母細(xì)胞壁,之后還需要蛋白酶K消化、苯酚-氯仿抽提等多步操作去除蛋白,最后經(jīng)過(guò)異丙醇沉淀和乙醇洗滌獲得染色體DNA,整個(gè)提取過(guò)程需要幾個(gè)小時(shí),且難以同時(shí)進(jìn)行批量操作。
本研究中直接從平板上刮取新鮮酵母菌體,省去液體培養(yǎng)、離心收集等步驟,可節(jié)約實(shí)驗(yàn)時(shí)間,減少工作量。研究中發(fā)現(xiàn)酵母的培養(yǎng)時(shí)間對(duì)染色體DNA的提取有較大影響,培養(yǎng)時(shí)間過(guò)長(zhǎng)不利于染色體DNA的提取。
本文建立的酵母染色體DNA快速提取方法僅需一步反應(yīng),且可同時(shí)處理多個(gè)樣品,所制備的染色體DNA無(wú)需任何純化處理即可作為模板用于酵母26S rDNA D1/D2區(qū)域序列的PCR擴(kuò)增,并獲得了良好的擴(kuò)增結(jié)果及擴(kuò)增產(chǎn)物的測(cè)序結(jié)果。研究中使用的酵母分屬于10個(gè)屬、17個(gè)種,證明建立的酵母染色體DNA快速提取方法具有廣泛的適用性。基于這種新的酵母染色體DNA提取方法可以實(shí)現(xiàn)大批量酵母的快速分子鑒定,本中心已將該方法應(yīng)用于國(guó)家微生物資源平臺(tái)的建設(shè),成功完成了800多株酵母分離物的分子鑒定工作。
[1] Barnett JA,Payne RW,Yarrow D.Yeasts:Characteristics and identification[M].UK:Cambridge University Press,2000.
[2] Deak T,Beuchat LR.Handbook of Food Spoilage Yeasts[M].USA:CRC Press,1996.
[3] Kurtzman C P,F(xiàn)ell J W.Yeast systematics and phylogenyimplications of molecular identification methods for studies in ecology.Biodiversity and Ecophysiology of Yeasts[M].Berlin:Springer-Verlag,2006.
[4] Bridge P D.The history and application of molecular mycology[J].Mycologist,2002,16:90 -99.
[5] Peterson S W,Kurtzman C P.Ribosomal R N A sequence divergence among sibling species of yeasts[J].Syst Appl Microbiol,1991,14:124 -129.
[6] Fell J W,Boekhout T,F(xiàn)onseca A,et al.Biodiversity and systematics of basidiomycetous yeasts as determined by large-subunit rDNA D1/D2 domain sequence analysis[J].Int J Syst Evol Microbiol,2000,50:1 351 -1 371.
[7] Kurtzman C P,Robnett C J.Identification and phylogeny of ascomycetous yeasts from analysis of nuclear large subunit(26S)ribosomal DNA partial sequences[J].Antonie van Leeuwenhoek,1998,73:331~371.
[8] Hierro N,González A,Mas A,et al.New PCR-based methods for yeast identification[J].J Appl Microbiol,2004,97(4):792~801.
[9] 周小玲,沈微,饒志明 等.一種快速提取真菌染色體DNA的方法[J].微生物學(xué)通報(bào),2004,31(4):89~92.
[10] Liu D,Coloe S,Baird R,et al.Rapid mini-preparation of fungal DNA for PCR[J].J Clin Microbiol.2000,38(1):471.
[11] Holm C,Meeks-Wagner D W,F(xiàn)angman W L,et al.A rapid,efficient method for isolating DNA from yeast[J].Gene,1986,42(2):169~173.
[12] Kurtzman C P,Robnett C J.Identification of clinically important ascomycetous yeasts based on nucleotide divergence in 5'end of the large subunit(26S)ribosomal DNA gene[J].J Clin Microbiol,1997,35:1 216 -1 223.
A Rapid Method for Yeast Molecular Identification
Chen Yuan-yuan1,2,Shi Gui-yang1,2,Wang Zheng-xiang1,2
1(The Key Laboratory of Industrial Biotechnology,Ministry of Education,Jiangnan University,Wuxi 214122,China)2(Culture& Information Center of Industrial Microorganism of China Universities,Jiangnan University,Wuxi 214122,China)
D1/D2 domain of 26S rDNA sequence-based alignment and analysis is a widely used method for yeast identification and classification.Rapid and repeatable protocols to extract and prepare the chromosomal DNA of yeast are the basic needs to acquire the templates for following amplification of D1/D2 domain.However,the present methods to extract chromosomal DNA are time-consuming and unable to deal with large-scale samples at the same time.In the present study,a novel rapid and efficient method to identify a large amount of yeast isolates in the same batch was established.The chromosomal DNA required only 20 minutes to prepare and can be directly used as template for PCR amplification.Results of amplified ITS sequences were clear and easy to identify by sequencing analysis.Key words yeast,chromosomal DNA,extraction,D1/D2 domain of 26S rDNA,identification
博士研究生(王正祥教授為通訊作者)。
*國(guó)家自然資源科技平臺(tái)(No.2005DKA21208)資助
2010-08-14,改回日期:2010-09-14
猜你喜歡
軍事文摘·科學(xué)少年(2021年1期)2021-02-04 08:03:45
中國(guó)科技博覽(2016年2期)2016-04-25 20:32:39
小學(xué)生導(dǎo)刊(2016年34期)2016-04-11 00:49:44
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
閱讀與作文(小學(xué)低年級(jí)版)(2015年8期)2015-05-30 10:48:04
小雪花·成長(zhǎng)指南(2015年4期)2015-05-19 14:47:56
電測(cè)與儀表(2015年5期)2015-04-09 11:30:52
食品工業(yè)科技(2014年9期)2014-03-11 18:15:31
食品科學(xué)(2013年19期)2013-03-11 18:27:43
