PA66/可反應性納米SiO2復合材料的非等溫結晶動力學
2011-12-04 08:47:10徐翔民葛玉萍張予東張治軍
中國塑料 2011年9期
關鍵詞:復合材料
徐翔民,葛玉萍,張予東,張治軍
(1.黃河水利職業技術學院機電工程系,河南 開封475004;2.河南大學特種功能材料教育部重點實驗室,河南 開封475004)
PA66/可反應性納米SiO2復合材料的非等溫結晶動力學
徐翔民1,2,葛玉萍1,張予東2,張治軍2
(1.黃河水利職業技術學院機電工程系,河南 開封475004;2.河南大學特種功能材料教育部重點實驗室,河南 開封475004)
通過雙螺桿擠出機制備出聚酰胺(PA)66/可反應性納米SiO2(RNS)復合材料,采用Jeziorny法和 Mo法對其非等溫結晶行為進行了研究。結果表明,RNS具有較強的異相成核能力,能提高PA66的結晶速率,并使PA66的晶體結構和生長機制發生改變;通過對比PA66及其復合材料的結晶活化能發現RNS能夠降低PA66的結晶活化能。
聚酰胺66;可反應性納米二氧化硅;復合材料;非等溫結晶
0 前言
PA66作為一種具有優良力學性能和良好可加工性的工程塑料,其改性研究一直受到廣泛關注[1-3]。近年來,隨著納米材料制備工藝的不斷發展和完善,納米材料在PA66中的應用研究開始逐漸升溫,但這方面的工作主要集中在 PA66/納米蒙脫土復合材料上[4-5]。對于目前在納米填料領域中使用量較大的RNS,其在PA66中的應用還少有報道。基于此,筆者對PA66/RNS復合材料進行了深入研究。在前期研究中[6],主要考察了PA66/RNS復合材料的力學性能和熱性能,發現復合材料的拉伸強度和彈性模量隨RNS含量的增加而提高。當RNS含量為3%(質量分數,下同)時,復合材料的拉伸強度達到最大,增幅為11%;彈性模量則在RNS含量為5%時達到最大,增幅為30%。不僅如此,加入RNS還使PA66的熱穩定性和熱動態力學性能得以大幅提高。
對于PA66這種半結晶聚合物,在納米填料加入后,所得復合材料的性能受較多因素的影響,不僅與基體/納米填料的界面結構有關,還與PA66的晶體結構和形態有關[7]。界面結構對PA66/RNS復合材料性能的影響在前文中已經討論,因此進一步的研究主要集中在晶體結構和形態對復合材料性能的影響上。當采用RNS對PA66進行填充改性時,PA66的結晶行為會發生改變,而結晶行為的改變會使PA66的晶體形態和晶粒尺寸發生變化,從而影響復合材料的性能。由此可以看出,加入RNS后,PA66結晶行為的變化情況將直接影響復合材料性能的改善。由于納米復合材料的制備基本上都是在非等溫條件下進行,那么考察非等溫條件下納米粒子對PA66結晶行為的影響就顯得非常有意義。
本文在制備出PA66/RNS復合材料后,首先通過透射電子顯微鏡和X射線衍射儀觀察和分析了復合材料的微觀結構,然后借助于差示掃描量熱儀對PA66/RNS復合材料的非等溫結晶行為進行了深入研究。
1 實驗部分
1.1 主要原料
PA66,EPR27,中國平頂山神馬集團;
RNS,河南大學河南省納米材料工程技術研究中心。
1.2 主要設備及儀器
雙螺桿擠出機,W50EHT,德國Brabender公司;
注射成型機,ES200/45,奧地利Engel公司;
紅外分析儀(FT-IR),AVATAR360,美國 Nicolet公司;
透射電子顯微鏡(TEM),JEM-2010,日本電子株式會社;
X射線衍射儀(XRD),X′Pert Pro,荷蘭Philips公司;
差示掃描量熱儀(DSC),DSC822e,瑞士Mettler公司。
1.3 樣品制備
RNS制備:通過原位表面修飾法[8],硅烷偶聯劑KH550為表面修飾劑,其形成機理如圖1所示。
PA66/RNS復合材料制備:所有原料在使用前均經過100℃真空干燥處理,然后將RNS填料與PA66切片分別按質量比1∶100、3∶100、5∶100混合均勻,并通過雙螺桿擠出機熔融共混擠出造粒,擠出溫度260~270℃,最后在注射成型機上制成標準樣條。
1.4 性能測試與結構表征
將一定量的復合材料試樣用甲酸溶解,并輔以超聲振蕩,待試樣溶解完全后,對溶液進行離心分離;得到的沉淀物再次用溶劑溶解,如此反復,直到離心液中檢測不到PA66溶解物;將最終分離出的RNS用甲醇洗滌并充分干燥后得到復合材料的抽提物,并對其進行FT-IR分析;
TEM分析:采用低溫切片機制樣,通過TEM觀察復合材料切片中RNS的分散情況,放大倍率為20000倍;
XRD分析:Cu靶,Kα射線,波長為0.15406nm,掃描速度為2(°)/min,測試范圍為3°~40°;
DSC分析:在N2氣氛中,將約5mg的樣品以20℃/min的速率升溫到300℃,恒溫5min以消除熱歷史,然后分別以5、10、20、40℃/min的速率降至室溫,記錄DSC曲線。
2 結果與討論
2.1 FT-IR分析
從圖2可以看出,在 RNS譜中,1390、1560cm-1處對應的是C—H伸縮振動峰和N—H彎曲振動峰,而2936cm-1峰的出現則說明有長的烷基鏈存在于RNS表面。對應圖1可知,RNS表面已經被改性,帶有氨基基團的烷基鏈被化學接枝到RNS表面。將PA66和PA66/RNS復合材料譜圖進行對比可以發現,PA66/RNS復合材料的譜圖中出現了與PA66相關的特征峰,如1640、1545cm-1處對應的伸縮振動峰及N—H彎曲振動峰,2940、2865cm-1處對應的C—H振動峰。特別是3310cm-1處N—H振動峰的出現(氫鍵)更說明了有一定量的PA66存在于RNS表面。由于PA66/RNS是經過反復抽提處理的,因此可以認為PA66鏈已經與RNS表面官能團發生了反應,并通過化學鍵合的方式接枝到RNS表面。
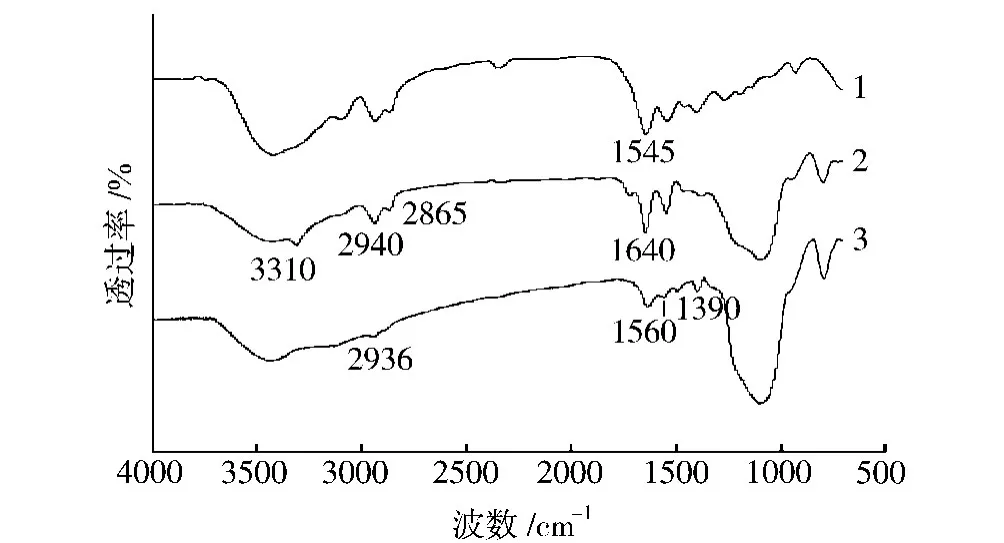
圖2 PA66、RNS及其復合材料的FT-IR譜圖Fig.2 FT-IR spectra for PA66,RNS and their composite
2.2 TEM與XRD分析
從圖3可以看出,RNS在PA66基體中分散較為均勻,但它主要是以團聚體形式出現,團聚體粒徑在100nm左右。RNS這一分散特征與其表面修飾結構及熔融共混擠出工藝有關。然而進一步的觀察發現,隨著納米RNS含量的增加,團聚體越來越多,其粒徑也在不斷增大,納米顆粒分散的均勻性降低。圖4表明純PA66在20.05°和23.01°處有2個強衍射峰(α1和α2),分別對應的是PA66的α晶的(100)面和(010)/(110)面。當RNS加入到PA66基體后,其所形成復合材料的XRD譜相對于純PA66有了明顯改變。首先,隨著RNS含量的增加,復合材料的α1和α2衍射峰逐漸向大角度方向移動,表明復合材料中α晶的晶面間距有所減小。其次,復合材料的XRD譜中出現了PA66的γ1結晶峰,且隨著RNS含量的增加,γ1結晶峰的強度逐漸增加。復合材料XRD譜的上述變化顯然與RNS的添加有關。對于PA66/RNS復合材料來說,RNS與PA66分子鏈之間大量氫鍵連接及化學鍵合的出現增加了PA66折疊鏈間的相互作用力,使α晶的晶面間距減小[9];而且強烈的界面相互作用還引起了PA66分子鏈排列結構的改變,導致γ晶的出現[10]。此外,RNS還使復合材料的α1衍射峰出現了一定程度的寬化,表明復合材料體系內PA66晶粒尺寸有所減小,晶粒的不完整程度增加。進一步對比α晶的衍射峰形狀還可發現,復合材料的α2衍射峰強隨著RNS含量的增加逐漸降低,而α1衍射峰強度則明顯增加。α2衍射峰強度的降低表明RNS能夠干擾PA66的α晶在(100)面(氫鍵平面)上的有序排列,使其優先沿(010)/(110)面生長,而α晶在氫鍵平面上有序排列受到干擾也是導致復合材料γ晶產生的主要原因。PA66/RNS復合材料中α1和α2衍射峰強的上述變化在PA66/黏土的研究中也被觀察到[11]。
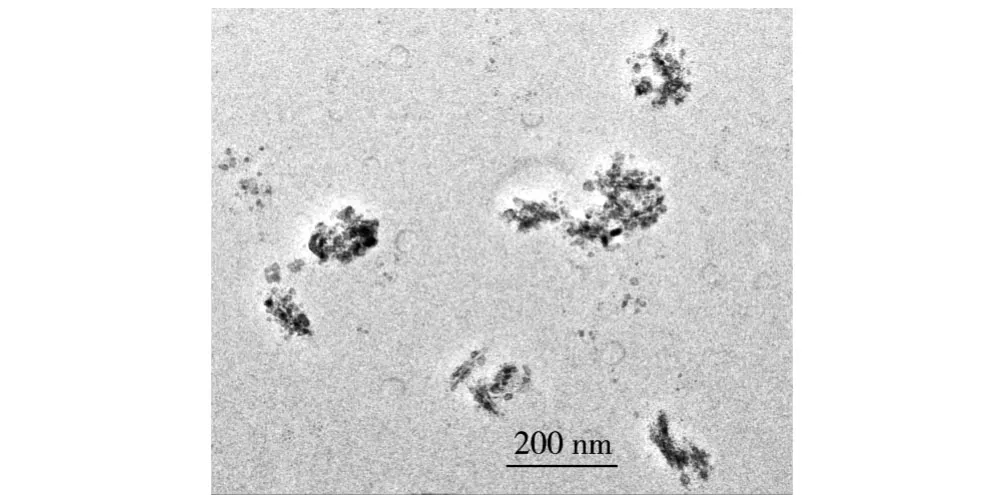
圖3 RNS含量為1%的復合材料的TEM形貌Fig.3 TEM micrograph for PA66/composite with 1%RNS
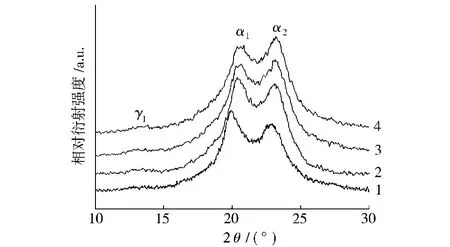
圖4 PA66及PA66/RNS復合材料的XRD譜Fig.4 X-ray diffraction patterns for PA66and PA66/RNS composites
2.3 PA66/RNS復合材料的非等溫結晶行為
從圖5可以看出,隨著降溫速率的增加,PA66復合材料結晶峰的位置逐漸向低溫方向移動,且峰的形狀也逐漸寬化。由此可見,降溫速率的增加使體系結晶過冷度增加,結晶溫度降低。這是因為隨著冷卻速率的不斷增加,PA66分子鏈規則排列進入晶格的能力逐漸減弱,結晶過程受阻,PA66需要較大的過冷度才能結晶。而在較低溫度下PA66的活動能力降低,導致結晶不完整程度增加,結晶峰隨之變寬。此外,對比結晶溫度隨降溫速率增加而下降的幅度還可以發現,當降溫速率從5℃/min提高到40℃/min時,PA66的結晶溫度下降了17.3℃,而復合材料的結晶溫度則下降了21.5℃,明顯高于PA66。這說明RNS對PA66鏈段的松弛及結晶過程有明顯的阻礙作用。這種阻礙作用可能源于RNS與基體PA66分子鏈之間強烈的界面相互作用。
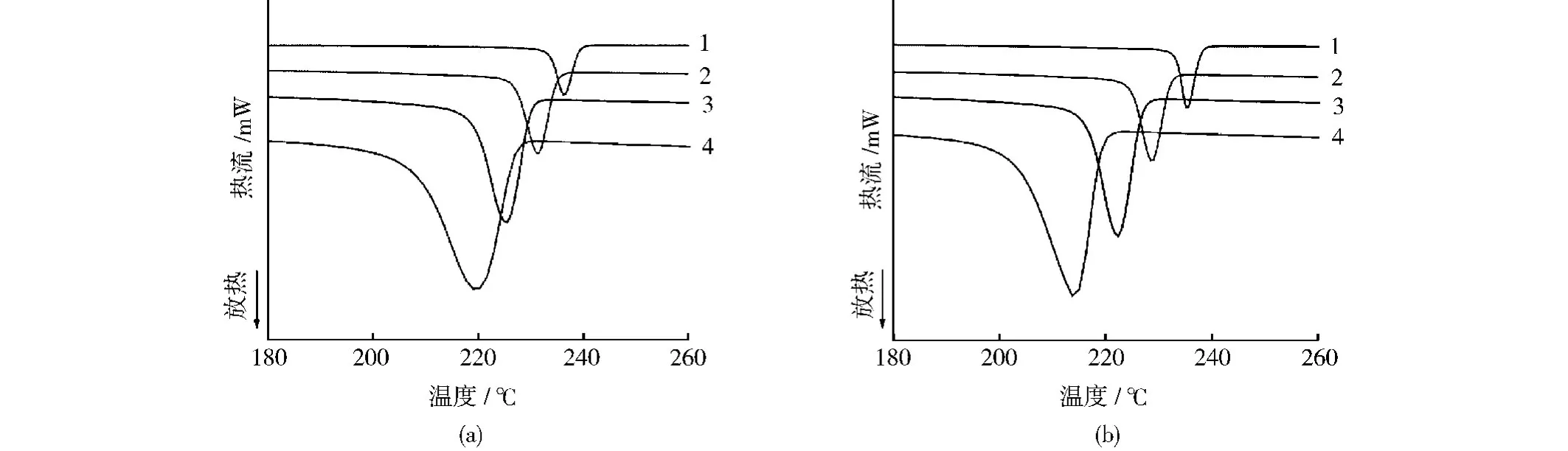
圖5 PA66及其復合材料的DSC結晶曲線Fig.5 DSC curves for PA66and its composites at different cooling rates
2.4 復合材料的非等溫結晶動力學
許多文獻報道了用DSC法研究聚合物非等溫結晶動力學[12-14],從處理等溫結晶的Avrami方程出發并考慮非等溫結晶的特點,對Avrami方程進行修正而得到一些處理非等溫結晶動力學的方法。本文采用的Jeziorny法和Mo法就是目前文獻中常處理非等溫結晶動力學的2種方法。
以Xt為任意結晶溫度(T)時的相對結晶度,二者之間存在下列關系:

式中 T0——結晶起始溫度,℃
T∞——結晶完成時的溫度,℃
d Hc——在有限溫度范圍dT中結晶所釋放出的焓值,J/g
據式(1)得到Xt與T 的關系,如圖6所示。隨著降溫速率的增加,PA66和復合材料達到同一相對結晶度所需的溫度明顯降低,且在相同降溫速率下,達到同一相對結晶度時PA66所對應的溫度要高于復合材料所對應的溫度。
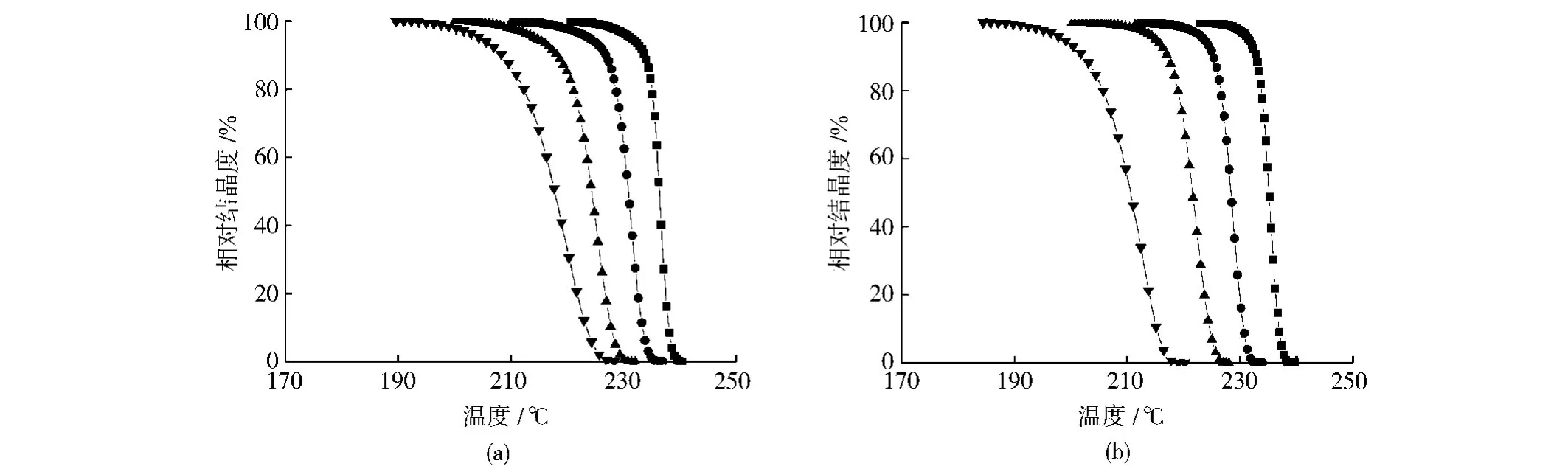
圖6 PA66及其復合材料的相對結晶度與溫度的關系曲線Fig.6 Curves for relative crystallinity of PA66and PA66/RNS composites with 1%RNS againsit temperature
分析等溫結晶動力學時最常用的方法是Avrami方程,它假定相對結晶度(Xt)與結晶時間(t)有以下關系:
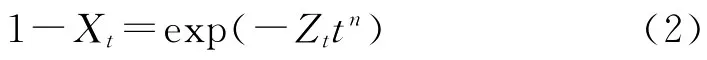
式中 n——Avrami指數
t——時間,s
Zt——結晶速率常數
Jeziorny法[12]就是將Avrami方程進行修正以用于處理非等溫結晶。先將式(2)兩邊同時取對數,得式(3):
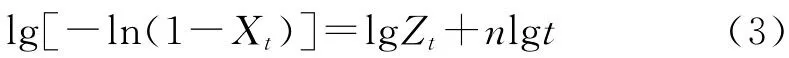
由于Avrami方程中Xt是t的函數,而通過DSC曲線得到的Xt為T的函數,因此這里需要對其進行時溫轉換:

式中 Ti——i時刻的結晶溫度,℃
Φ——降溫速率,℃/min
圖7給出了PA66和復合材料的Xt與t之間的相互關系,通過此圖可得到PA66和復合材料的半結晶時間(t1/2),其結果列于表2中。從圖8可以看出,在結晶初期,PA66與復合材料的lg[-ln(1-Xt)]與lgt保持著較好的線性關系,但在結晶后期,由于晶粒長大,并發生相互碰撞,限制了晶體的自由生長,使曲線出現偏離。由圖8中曲線線性部分的斜率和截距可以分別求出n和Zt。考慮到非等溫結晶特點,Jeziorny認為Zt應該被修正,降溫速率Φ的因素應該被考慮。假設Φ為常數或大致為常數,那么非等溫結晶速率常數Zc的最終形式應為:


圖7 PA66及其復合材料的相對結晶度與時間的關系Fig.7 Curves for relative crystallinity for PA66and PA66/RNS composite with 1%RNS vs time
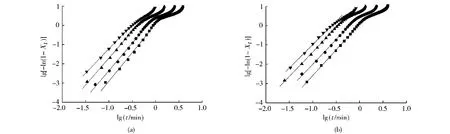
圖8 PA66及其復合材料的lg[-ln(1-Xt)]與lgt的關系Fig.8 Plots of lg[-ln(1-Xt)]and lgt for PA66and PA66/RNS composite with 1%RNS
在非等溫條件下,n反映的是聚合物一次結晶時的晶體生長維數,且n值越大,晶體越完善[15]。由表1可以看出,隨著Φ的增加,PA66與復合材料的n值依次出現了不同程度的降低,表明晶體的完善程度也在不斷降低。純PA66的n值平均為2.88,接近3,而復合材料的n值平均為2.54。由于純PA66制備過程中有其他助劑的加入,有助于PA66異相成核,這種情況下n值接近3,表明純PA66中晶體生長主要是以三維生長方式為主。對復合材料來說,其n值相對于純PA66有明顯降低,表明RNS對PA66的結晶行為產生了較大影響,RNS的加入進一步降低了PA66晶體的完整程度,同時也使PA66晶體的生長方式由三維生長逐漸向二維生長過渡。此外,由表1還可以看出,無論是純PA66,還是復合材料,其半結晶時間t1/2隨著Φ的增加不斷減小,而Zc則是不斷增大,但當Φ達到40℃/min時,二者的Zc都出現了一定程度的降低。這是因為降溫速率的提高使體系過冷度提高,而高的過冷度可提高PA66的成核率,大量成核點的出現減少了PA66鏈段的運動距離。成核點的增多,運動距離的縮短使PA66的結晶速率增加,半結晶時間縮短。當Φ過高時,鏈段運動及晶格重排嚴重受阻,制約了PA66的結晶,使體系結晶速率降低。在同一冷卻速率下,復合材料的Zc高于純 PA66,t1/2也較 PA66有所減小,說明RNS對PA66具有良好的異相成核能力。
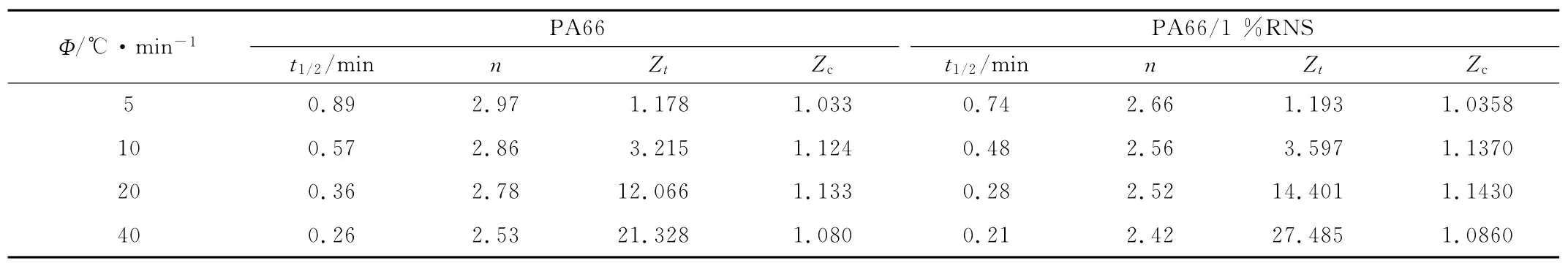
表1 PA66及其復合材料的非等溫結晶動力學參數Tab.1 Non-isonthermal kinetics parameters at different relative crystallinity of PA66and PA66/RNS composites with 1%RNS
考慮到降溫速率對結晶過程的影響,Ozawa[13]將Avrami方程推廣到非等溫結晶過程中:
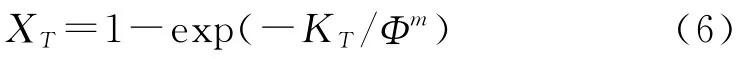
式中 XT——T溫度時的相對結晶度,%
m——Ozawa指數
KT——降溫函數
對式(6)兩次取對數,得到:
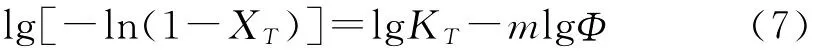
根據斜率m和截距lgKT進行非等溫結晶動力學研究,但式(7)只適用于降溫速率較小的情況。
莫志深等[14]結合了Avrami方程和Ozawa方程,將Φ和t聯系起來,得到式(8):
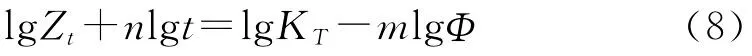
進行整理后得式(9):

式中 F(T)——單位結晶時間達到一定結晶度所需的降 溫 速 率,F(T)= [KT/Zt]1/m,℃/min
a——Avrami指數n與Ozawa指數m之比,即a=n/m
從圖9可以看出,PA66與復合材料均具有非常好的線性關系,這說明用Mo方法處理非等溫結晶過程是可行的,由直線斜率和截距可分別求出a和F(T),觀察表2可以發現,樣品的a變化不大,F(T)隨結晶度的增加而增加,表明在相同時間內,同一種材料要達到的相對結晶度越大,其所需的冷卻速率也就越大。在相同結晶度下,復合材料的F(T)比純PA66小,表明在相同的時間內達到同一結晶度時復合材料所需的降溫速率較小,也就是說復合材料的結晶速度要高于純PA66,這一結果與Jeziorny法處理所得結果相一致。
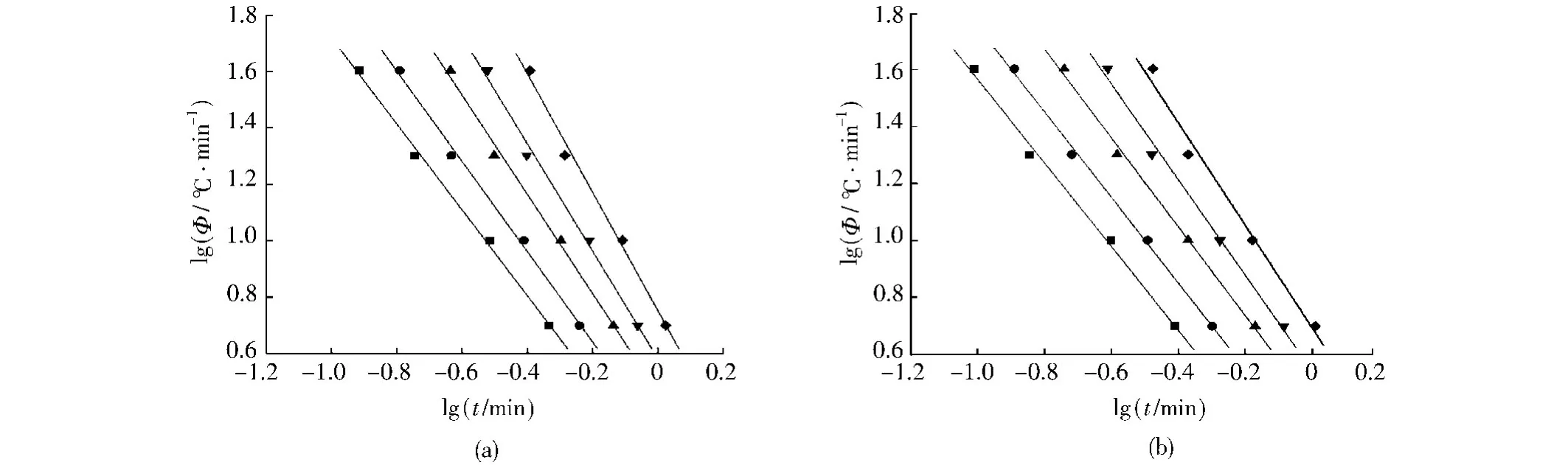
圖9 PA66及其復合材料的lgt與lgΦ的關系Fig.9 Plots of lgΦfor PA66and PA66/RNS composite with 1%RNS against lgt
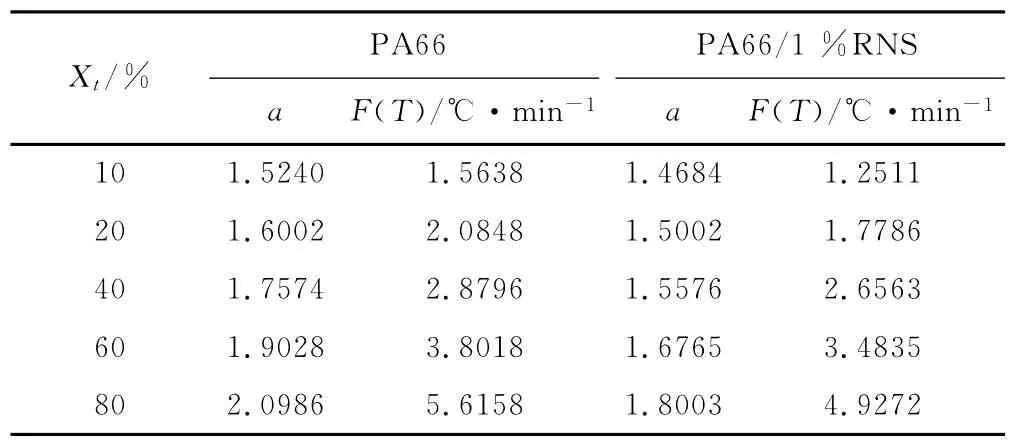
表2 PA66及復合材料Mo法的非等溫結晶動力學參數Tab.2 Non-isothermal crystallization kinetics parameters of PA66and PA66/RNS with 1%RNS from Mo equation
2.5 復合材料的結晶活化能
Kissinger[16]提出了結晶活化能可由結晶峰溫隨降溫速率變化來決定,其表達式為:
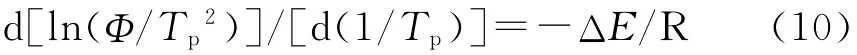
式中 Tp——結晶峰溫度,℃
ΔE——結晶活化能,kJ/mol
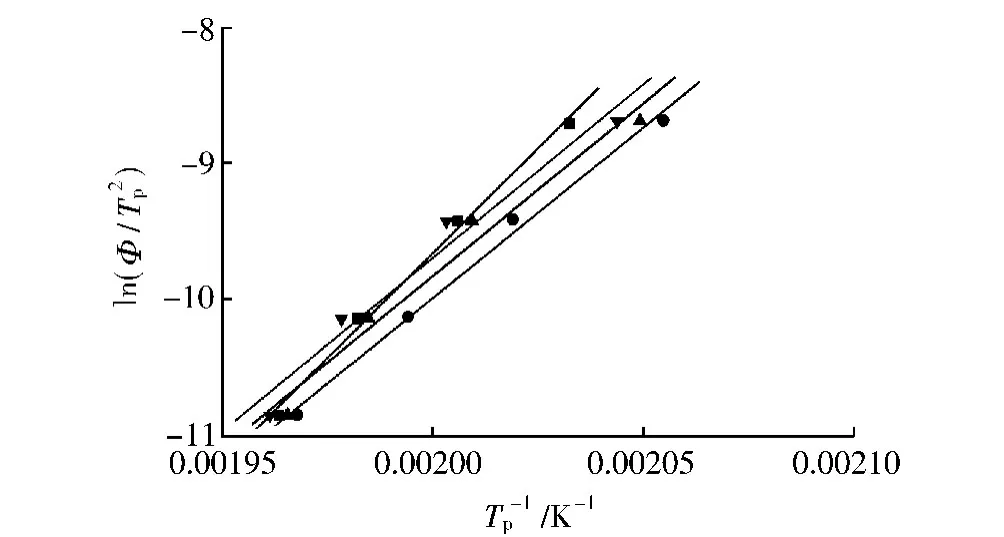
圖10 PA66及其復合材料的ln(Φ/Tp2)和1/Tp之間的關系Fig.10 Plots for ln(Φ/Tp2)of neat PA66 and its composites versus 1/Tp
圖10給出了PA66及其復合材料的ln(Φ/Tp2)~1/Tp的關系,通過線性擬合,結晶活化能ΔE能夠通過直線的斜率計算得出。這里分別計算了RNS含量為0、1%、3%、5%的復合材料的ΔE,其值大小分別為256.2、208.5、211.0、211.9kJ/mol。顯然,RNS的加入降低了PA66的ΔE。由于ΔE包括初始成核自由能和熔體-晶體轉換自由能。RNS與PA66之間強烈的界面作用,使PA66在RNS周圍發生結晶時鏈段排列所需能量降低,即復合材料相對于純PA66降低的那部分結晶活化能由RNS與PA66的作用能提供,反映出RNS在PA66結晶過程中的異相成核作用[17]。RNS的這種異相成核能力降低PA66的初始成核自由能,提高PA66的成核率;但RNS與PA66鏈之間的相互作用也會影響PA66分子鏈的運動,阻礙PA66晶粒的生長,反過來會引起熔體-晶體轉換自由能增加。因此,ΔE的大小取決于二者的綜合作用。本研究中,復合材料ΔE低于純PA66這一結果表明,初始成核自由能的降低對結晶活化能的影響較大。值得注意的是,初始成核自由能的降低,即RNS的異相成核作用,并沒有帶來復合材料結晶溫度的升高,這是因為RNS雖然可以使PA66在較高溫度下成核,但成核率較低,強烈的鏈段阻礙作用以及較長的運動距離限制了PA66的結晶。隨著溫度的降低,體系過冷度不斷增加,復合材料的異相成核點也在不斷增多,縮短了PA66鏈段運動的距離,有效地減少了RNS對PA66鏈段運動的阻礙,PA66的結晶速率因此大幅上升,結晶過程快速完成。綜上所述,盡管RNS具有較好的異相成核能力,但其對PA66鏈段運動較強的阻礙作用,使得復合材料中PA66結晶仍需一定的過冷度才能順利完成,這也就成為復合材料結晶溫度降低的主要原因。
3 結論
(1)RNS通過熔融共混的方式加入到PA66基體后,其表面官能團與PA66鏈發生強烈的界面作用,使PA66鏈段以化學鍵合的方式接枝到RNS表面;
(2)RNS與PA66之間的強相互作用引起PA66晶體結構的改變,導致γ晶的產生;RNS的存在還限制了PA66晶粒的生長,使晶粒尺寸減小,晶粒不完整度增加。隨著降溫速率的增加,PA66及其復合材料的結晶峰不斷變寬,結晶溫度逐漸向低溫方向移動,顯示出RNS對PA66結晶的阻礙作用;
(3)Jeziorny法和Mo法均適合處理PA66及其復合材料的非等溫結晶過程。通過這兩種方法分析發現,RNS能夠降低PA66晶體的完善程度,加快結晶速率,縮短半結晶時間,并使PA66晶體生長方式由三維生長向二維生長過渡;
(4)RNS的加入降低了PA66的ΔE,表明RNS具有良好的異相成核能力,能夠加快PA66晶核形成。
[1] Zou H,Zhang Q,Tan H,et al.Clay Locked Phase Morphology in the PPS/PA66/Clay Blends During Compounding in an Internal Mixer[J].Polymer,2006,47(1):6-11.
[2] Lu Y L,Zhang Y,Zhang G B,et al.Influence of Thermal Processing on the Perfection of Crystals in Polyamide 66and Polyamide 66/Clay Nanocomposites[J].Polymer,2004,45(26):8999-9009.
[3] Mehrabzadeh M,Kamal M R.Melt Processing of PA-66/Clay,HDPE/Clay and HDPE/PA-66/Clay Nanocomposites[J].Polymer Engineering and Science,2004,44(6):1152-1161.
[4] Shen L,Phang I Y,Liu T X,et al.Nanoindentation and Morphological Studies on Nylon 66/Organoclay Nanocomposites.II.Effect of Strain Rate[J].Polymer,2004,45(24):8221-8229.
[5] Han B,Ji G D,Wu S S,et al.Preparation and Characterization of Nylon 66/Montmorillonite Nanocomposites with Co-treated Montmorillonites[J].European Polymer Journal,2003,39(8):1641-1646.
[6] 徐翔民,張予東,李賓杰,等.納米SiO2/尼龍66復合材料的力學性能和熱性能[J].復合材料學報,2008,25(4):56-61.
[7] Jordan J,Jacob K I,Tannenbaum R,et al.Experimental Trends in Polymer Nanocomposites—A Review[J].Materials Science and Engineering A,2005,393(1/2):1-11.
[8] Li X H,Cao Z,Zhang Z J,et al.Surface-modification In Situ of Nano-SiO2and Its Structure and Tribological Properties[J].Applied Surface Science,2006,252(22):7856-7861.
[9] Wang H L,Shi T J,Yang S Z,et al.Crystallization Behavior of PA66/SiO2Organic-inorganic Hybrid Material[J].Journal of Applied Polymer Science,2005,101(2):810-817.
[10] Lincoln D M,Vaia R A,Wang Z G,et al.Temperature Dependence of Polymer Crystalline Morphology in Nylon 6 Montmorillonite Nanocomposites[J].Polymer,2001,42(25):9975-9985.
[11] Liu X H,Wu Q J,Berglund L A.Polymorphism in Polyamide 66/Clay Nanocomposites[J].Polymer,2002,43(18):4967-4972.
[12] Jeziorny A.Parameters Characterizing the Kinetics of the Non-isothermal Crystallization of Poly(ethylene terephthalate)Determined by DSC[J].Polymer,1978,19(10):1142-1144.
[13] Ozawa T.Kinetics of Non-isothermal Crystallization[J].Polymer,1971,12(3):150-158.
[14] Liu T X,Mo Z S,Wang S E,et al.Nonisothermal Melt and Cold Crystallization Kinetics of Poly(aryl ether ether ketone ketone)[J].Polymer Engineering and Science,1997,37(3):568-575.
[15] Xu G X ,Lin S G.Study on the Non-isothermal Crystallization Kinetics of Syndiotactic Polystyrene(sps)[J].Polymer Materials Science & Engineering,1999,15(2):65-68.
[16] Kissinger H E.Reaction Kinetics in Differential Thermal Analysis[J].Analytical Chemistry,1957,29(11):1702-1706.
[17] 徐衛兵,戈明亮,何平笙.聚丙烯/蒙脫土納米復合材料非等溫結晶動力學的研究[J].高分子學報,2001,(5):584-587.
Non-isothermal Crystallization Kinetics of PA66/Reactable Nano-SiO2Composites
XU Xiangmin1,2,GE Yuping1,ZHANG Yudong2,ZHANG Zhijun2
(1.Department of Mechanical and Electrical Engineering,Yellow River Conservancy Institute,Kaifeng 475004,China;2.Key Lab for Special Functional Materials,Henan University,Kaifeng 475004,China)
PA66/reactable nano-SiO2composites were prepared in a twin-screws extruder by melt compounding.The non-isothermal crystallization of the composites and neat PA66was studied through Jeziorny and Mo methods based on DSC analysis.It showed that reactable nano-SiO2had a strong heterogeneous nucleation effect on PA66matrix.Its addition increased the crystallization rate of PA66,and led to changes in crystal structure,growth mechanism,and activation energy of PA66.
polyamide 66;reactable nano-silca;composite;non-isothermal crystallization
TQ323.6
B
1001-9278(2011)09-0048-08
2011-05-12
高等學校博士學科點專項科研基金資助項目(20060475001)
聯系人,xxm326@yahoo.com.cn
猜你喜歡
建材發展導向(2022年2期)2022-03-08 01:44:04
建材發展導向(2021年14期)2021-08-23 00:56:16
中國材料進展(2019年10期)2019-12-07 05:32:14
纖維復合材料(2018年3期)2018-04-25 07:22:58
電子測試(2017年11期)2017-12-15 08:57:13
山東工業技術(2016年15期)2016-12-01 05:31:34
中國塑料(2015年6期)2015-11-13 03:02:54
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年8期)2015-10-14 01:10:41
應用化工(2014年10期)2014-08-16 13:11:29
