復方歸芍婦康膠囊的制備與質量控制
2011-11-06 11:11:24張霞江延輝
中國現代中藥 2011年8期
張霞,江延輝
(哈爾濱星火藥物研究院,黑龍江 哈爾濱 150060)
復方歸芍婦康膠囊的制備與質量控制
張霞*,江延輝
(哈爾濱星火藥物研究院,黑龍江 哈爾濱 150060)
目的:制備復方歸芍婦康膠囊,建立其質量控制方法。方法:采用薄層色譜法對當歸和益母草進行定性鑒別,采用HPLC對復方歸芍婦康膠囊中的芍藥苷進行含量測定。結果:芍藥苷在0.303~1.515μg與峰面積呈良好的線性關系(r=0.999 8);平均回收率為99.66%,RSD=0.27%(n=6)。結論:該制劑制備工藝穩定,含量測定方法準確可靠,重現性好,可用于復方歸芍婦康膠囊的質量控制。
復方歸芍婦康膠囊;制備;芍藥苷;HPLC
復方歸芍婦康膠囊由當歸、白芍、益母草、熟地黃、白術、甘草等中藥組成,具有活血化瘀、通經止痛之功效,用于月經不調,經期疼痛等癥。為了更好地控制其內在質量,對處方中的當歸、益母草進行了定性鑒別分析,采用高效液相色譜法測定君藥白芍中芍藥苷的含量。
1 儀器及試藥
1.1 儀器
TN-200多功能中藥提取濃縮機組;日本島津LC-10AT液相色譜儀;SPD-10AVP紫外檢測器;硅膠G薄層板(自制)。
1.2 試藥
芍藥苷對照品(中國藥品生物制品檢定所,批號:110736-200912);當歸對照藥材(中國藥品生物制品檢定所,批號:120927-201014);鹽酸水蘇堿對照品(中國藥品生物制品檢定所,批號:110712-201010),乙腈為色譜純;正己烷、醋酸乙酯、正丁醇、鹽酸、乙醇等試劑均為分析純。
2 處方與制備
2.1 處方組成
當歸、白芍、益母草、熟地黃、白術、甘草6味中藥。
2.2 制備
取當歸、白芍、益母草、熟地黃、白術、甘草6味中藥加水煎煮3次,每次2 h,合并煎液,濾過,濾液濃縮至清膏,60℃干燥,粉碎,加淀粉、羧甲淀粉鈉適量,混勻,以50%乙醇為黏合劑制軟材,搖擺制粒機制粒,干燥,整粒,灌入硬膠囊,即得。批號:20101201,20101202,20101203。
3 質量控制
3.1 性狀
本品為硬膠囊,內容物為棕黃色顆粒或粉末,微苦。
3.2 鑒別
3.2.1 當歸的薄層鑒別[1]取本品內容物3 g,研細,加乙醚30 mL,加熱回流40 min,濾過,濾液揮干,殘渣加醋酸乙酯1 mL使溶解,作為供試品溶液。另取當歸對照藥材0.2 g,加乙醚10 mL,同法制成對照藥材溶液。再按處方量及制備工藝要求制成不含當歸的陰性對照樣品,同法制成陰性對照溶液。吸取上述3種溶液各2μL,分別點于同一硅膠G薄層板上,以正己烷-醋酸乙酯(9∶1)為展開劑,展開,取出,晾干,置紫外光燈(365 nm)下檢視。供試品色譜中在與對照藥材色譜相應的位置上,顯相同顏色的熒光斑點,見圖1。

圖1 當歸薄層色譜圖
3.2.2 益母草的薄層鑒別[2]取本品內容物5 g,加乙醇50mL,超聲15min,濾過,濾液置水浴上濃縮至約5 mL,加無水乙醇50 mL,攪拌,濾過,濾液置水浴上濃縮至約1mL,作為供試品溶液。另取鹽酸水蘇堿對照品加無水乙醇制成含lmg·mL-1的溶液,作為對照品溶液。按處方量及制備工藝要求制成不含益母草的陰性對照樣品,同法制成陰性對照溶液。吸取上述3種溶液各5μL,分別點于同一硅膠G薄層板上,以正丁醇-鹽酸-醋酸乙酯(8∶3∶1)為展開劑,展開,取出,晾干,噴以稀碘化鉍鉀試液,結果供試品色譜中在與對照品色譜相應位置上顯相同的橙紅色斑點,陰性對照無干擾,見圖2。
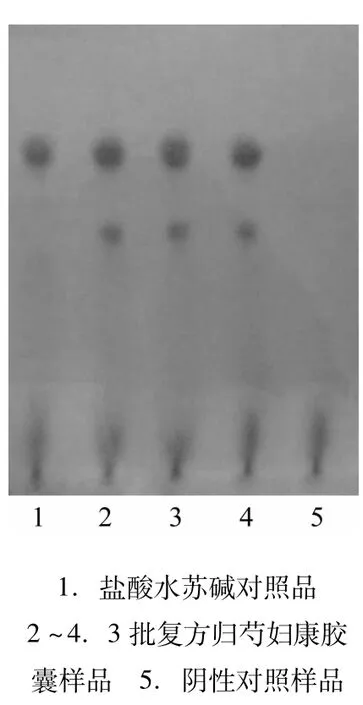
圖2 益母草薄層色譜圖
3.3 含量測定
3.3.1 色譜條件[3]色譜柱:Inertsil ODS-3不銹鋼色譜柱(4.6 mm×250 mm,5μm),流動相:磷酸0.1%溶液-乙腈(82∶18),流速:1.0 mL·min-1,柱溫:30℃,測定波長:230 nm,理論板數按芍藥苷峰計算應不低于3 000。
3.3.2 對照品溶液的制備 精密稱定芍藥苷對照品15.15 mg,置50 mL量瓶中,加甲醇溶解并稀釋至刻度,搖勻,即得(每lmL含0.303 mg芍藥苷)。
3.3.3 供試品溶液的制備 取本品10粒,研細,取約1.0 g,精密稱定,置具塞錐形瓶中,精密加入甲醇20 mL,密塞,稱定重量,超聲處理30 min,放冷,再稱定重量,用甲醇補足減失的重量,濾過,取續濾液作為供試品溶液。
3.3.4 系統適用性試驗 按處方比例和制法,制成不含白芍的樣品,并按供試品溶液的制備法制成空白對照溶液。依法進樣測定,結果空白對照溶液在芍藥苷保留時間處無吸收峰。表明在此實驗條件下,其他藥材成分對測定無干擾。見圖3。
3.3.5 標準曲線的繪制 精密取芍藥苷對照品溶液,分別吸取1.0,2.0,3.0,4.0,5.0 mL置10mL量瓶中,分別加甲醇稀釋至刻度,分別吸取10μL進樣,測定芍藥苷峰面積,以峰面積為縱坐標,進樣量為橫坐標進行線性回歸,求得回歸方程為Y=532 654.36X-29 513.24;r=0.999 8,結果表明芍藥苷在0.303~1.515μg線性關系良好。
3.3.6 穩定性試驗 取本品適量,制成供試品溶液,分別于0,2,4,6 h測定,對芍藥苷穩定性進行考察,結果RSD=0.37%,表明芍藥苷在6 h內基本穩定,能夠滿足測定要求。
3.3.7 重復性試驗 分別取本品各5份,制備供試品溶液,測定,結果RSD=0.41%,表明重現性較好,說明測量方法準確可靠。
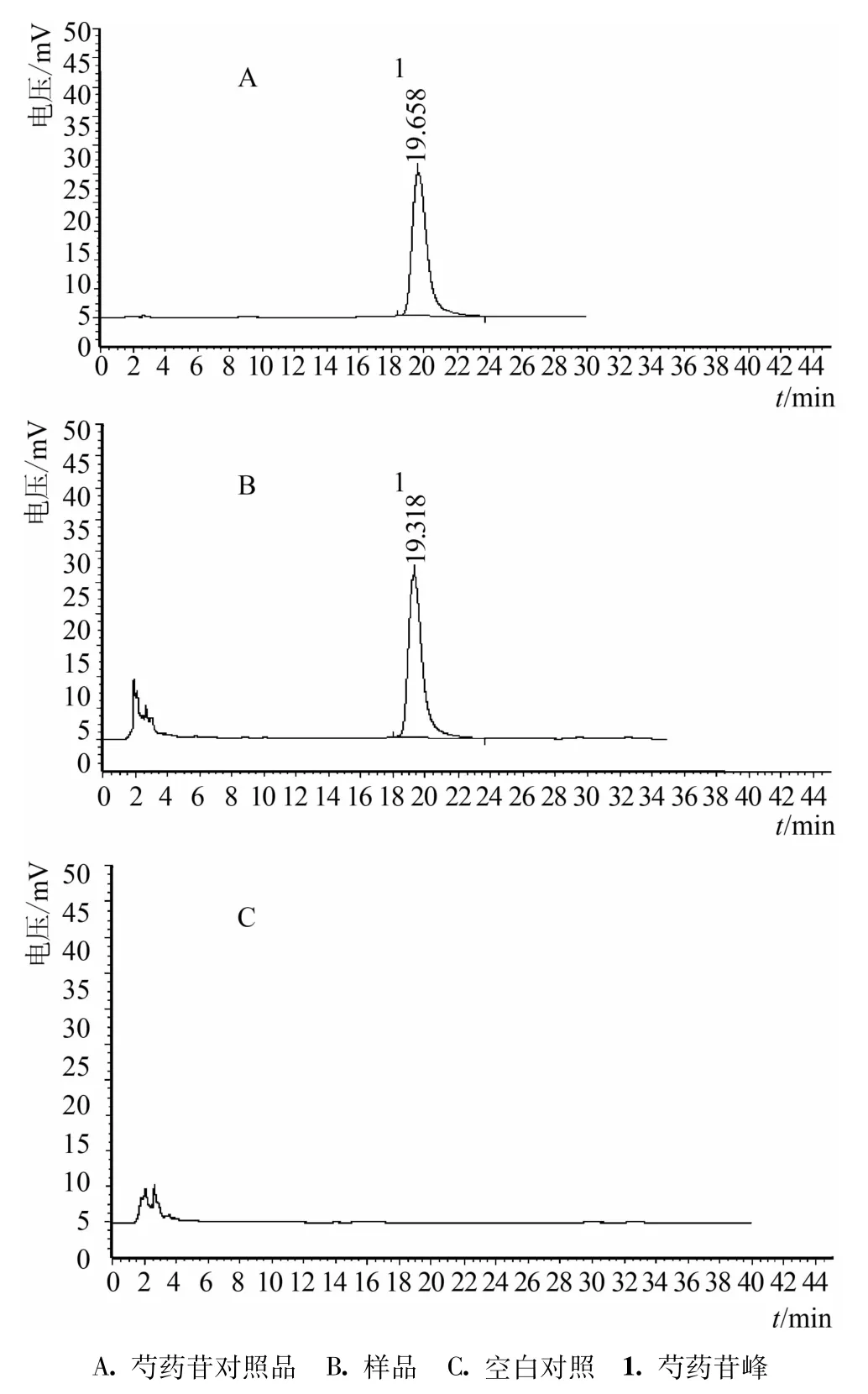
圖3 復方歸芍婦康膠囊樣品及對照品HPLC圖
3.3.8 精密度試驗 取樣品適量,制備供試品溶液,進樣6次,計算RSD=0.39%,結果表明該方法精密度良好。
3.3.9 加樣回收率試驗 取已知芍藥苷含量的樣品(批號:20101201,0.821 mg·g-1),精密稱取 6份,分別精密加入芍藥苷對照品適量,按樣品測定項下方法測定芍藥苷的量,計算回收率,結果見表1。
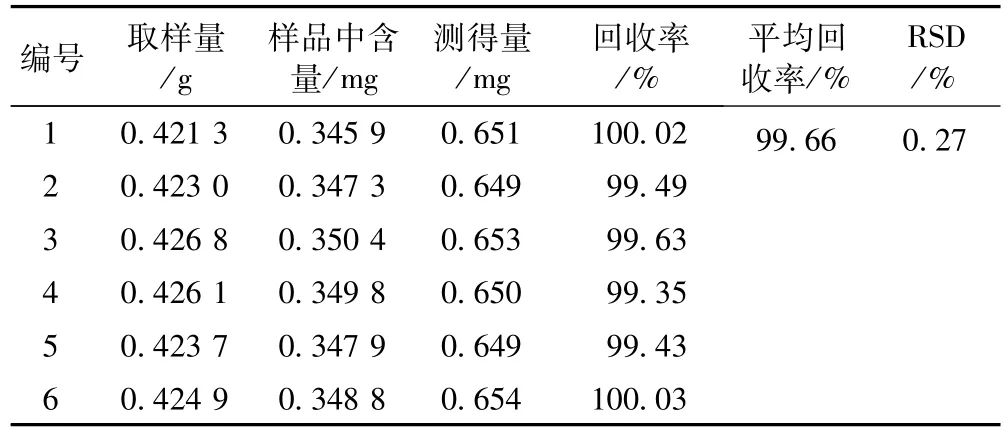
表1 芍藥苷回收率試驗
3.3.10 樣品含量測定 取3批樣品,分別按供試液制備方法制備。注入色譜儀,測定3批樣品中芍藥苷含量,結果見表2。

表2 復方歸芍婦康膠囊樣品中芍藥苷的含量測定
4 討論
本文通過實驗,確定了復方歸芍婦康膠囊中當歸、益母草的薄層色譜鑒別方法,陰性對照表明其他成分對本法無干擾,說明該方法專屬性較強,結果可靠。采用高效液相色譜法測定樣品中芍藥苷的含量,取得了較好的分離效果,經線性考察、系統適用性試驗、方法精密度考察、穩定性考察均良好,平均加樣回收率為99.66%,實驗方法簡便快速,適用于復方歸芍婦康膠囊中芍藥苷的含量測定。
[1]李霽明.TLC法鑒別頸椎活血膠囊中當歸、川芎、三七和人工牛黃等成分[J].安徽醫藥,2007,11(1):281.
[2]郝立芳.益母草沖劑中益母草的薄層色譜鑒別法[J].海峽藥學,2004,16(4):89-90.
[3]雷黎明,王先教,周家茂,等.活絡健骨膠囊中芍藥苷成分的測定方法研究[J].湖南環境生物職業技術學院學報,2010,16(2):25-28.
Preparation and Quality Control of Compound Guishao Fukang Capsules
ZHANG Xia,JIANG Yan-hui
(HarbinXinghuoPharmaceuticalResearchAcademy,Harbin150060,China)
Objective:To prepare compound Guishao Fukang Capsules and establish its quality controlmethod.Methods:TLCwas used to identifyAngelicasinensisandHerbaleonuri,HPLCwas used to determine the content of paeoniflorin in compound Guishao Fukang Capsules.Results:The linear ranges of paeoniflorin was 0.303~1.515μg(r=0.999 8),the average recovery was 99.66%,RSD was 0.27%(n=6).Conclusion:The preparation was stability,the content determination method was accurate reliable and reproducible and can be used in the quality control of compound Guishao Fukang Capsules.
Compound Guishao Fukang Capsules;Preparation;Paeoniflorin;HPLC
*張霞,(0451)86813688,E-mail:may1016@126.com
2011-05-03)
