TCIRG1基因突變致常染色體隱性遺傳性骨硬化癥的臨床表現及家系分析
2011-06-13 01:02:54何進衛傅文貞顧潔梅胡偉偉柯耀華胡云秋劉玉娟章振林
中國全科醫學 2011年35期
關鍵詞:基因突變
汪 純,何進衛,傅文貞,張 浩,顧潔梅,胡偉偉,岳 華,柯耀華,胡云秋,李 淼,劉玉娟,章振林
TCIRG1基因突變致常染色體隱性遺傳性骨硬化癥的臨床表現及家系分析
汪 純,何進衛,傅文貞,張 浩,顧潔梅,胡偉偉,岳 華,柯耀華,胡云秋,李 淼,劉玉娟,章振林
目的 通過對1例常染色體隱性遺傳性骨硬化癥 (ARO)患兒臨床表型和致病基因的研究,提高臨床醫師對這類少見惡性遺傳性疾病的認識。方法 對此例ARO患兒進行生化指標檢測、腹部超聲和骨骼X線檢查,同時對本例患兒及其父母、祖父母和外祖父母進行T細胞免疫調節子1基因 (TCIRG1)全編碼外顯子測序,并以100位健康志愿者作為基因突變分析的對照。結果 該例患兒血常規示血紅蛋白74 g/L,血小板24×109/L;血清堿性磷酸酶(ALP)達1 115 U/L;腹部超聲檢查示肝脾腫大,上肢和胸部X線可見典型的骨密度增高。患兒及其母親和外祖母均為TCIRG1基因3號外顯子第78位核苷酸發生雜合移碼突變 (3705C/-)攜帶者。而在100例健康者中未檢出該突變。結論 新發現的TCIRG1基因外顯子3雜合移碼突變 (3705C/-)是本例ARO患兒的致病突變位點,此基因突變導致脾腫大、嚴重貧血和血小板減少等嚴重的臨床表現,因此基因檢測對于診斷骨硬化癥至關重要。
骨硬化癥;遺傳性疾病,先天性;T細胞免疫調節子1基因
骨硬化癥是以破骨細胞缺乏或功能缺陷導致骨吸收障礙和高骨密度為臨床特征的遺傳性疾病。按照遺傳方式可分為常染色體顯性遺傳骨硬化癥 (autosomal dominant osteopetrosis,ADO)和常染色體隱性遺傳骨硬化癥 (autosomal recessive osteopetrosis,ARO)。其中ARO的新生兒發病率為1:250 000,主要表現為幼年起病,伴有嚴重且惡性的臨床癥狀,包括危及生命的骨髓衰竭、肝脾腫大、生長遲緩和神經系統缺陷 (失明和耳聾等)。目前已經明確ARO的主要致病基因是編碼破骨細胞特異性空泡型質子泵a3亞單位的T細胞免疫調節子1基因 (T - cell immune regulator 1 gene,TCIRG1)[1]、氯化物通道7基因 (Chloride channel 7,CLCN7)和骨硬化相關跨膜蛋白1基因 (Osteopetrosis-associated transmembrane protein 1,OSTM1)[2-5]。迄今中國有關ARO的報道僅見2007年由香港研究者報道的1例由CLCN7基因純合突變 (p.I261F)導致的ARO患兒[6]。本文通過對1例由TCIRG1基因雜合突變導致的ARO患兒的報道,闡述其典型臨床表現和致病基因,旨在提高臨床醫師對這類少見的惡性遺傳性疾病的了解。
1 病例簡介
患兒,女,6月齡,足月剖宮產,出生時體質量3 100 g。因嚴重貧血和腹部包塊在上海交通大學附屬第六人民醫院骨質疏松和骨病專科就診,父母為非近親結婚,家族中無類似發病。
指標檢測:接診后對該患兒進行了生化指標檢測、骨髓穿刺、腹部超聲和X線檢查。采集該患兒及其父母、祖父母和外祖父母的外周血并從中抽提基因組DNA,進行TCIRG1基因全編碼外顯子測序,共設計了10對引物,擴增了10個350~700 bp片段,覆蓋了該基因19個編碼外顯子,上下游引物序列見表1。同時以100位健康志愿者作為基因突變分析的健康對照[7]。

表1 TCIRG1基因全編碼外顯子測序上下游引物序列Table 1 The forward and reverse primers for the whole exon sequencing in TCIRG1 gene
臨床癥狀和致病基因:該患兒血常規示血紅蛋白74 g/L,血小板24×109/L;血清堿性磷酸酶 (ALP)1 115 U/L(同年齡參考值為115~460 U/L),天門冬氨酸氨基轉移酶 (AST)701 U/L(同年齡參考值為18~74 U/L),乳酸脫氫酶 (LDH)695 U/L(同年齡參考值為150~360 U/L),羥丁酸脫氫酶(HBDH)484 U/L(同年齡參考值為72~182 U/L),血鈣2.12 mmol/L(同年齡參考值為2.20~2.70 mmol/L),血磷0.96 mmol/L(同年齡參考值為1.23~2.00 mmol/)。腹部超聲檢查示肝、脾腫大。上肢和胸部X線可見典型的骨密度普遍增高,但無典型三明治樣椎體和骨內骨 (bone-in-bone)表現 (見圖1)。骨髓穿刺檢查示粒系增生輕度減低。患兒及其母親和外祖母均發現TCIRG1基因3號外顯子78位核苷酸發生雜合移碼突變 (3705C/-)導致91位蛋白質之后的氨基酸編碼發生改變 (見圖2),而患兒的父親和外祖父均為正常野生型,家系圖見圖3。在100例健康對照者中未發現此基因突變。
診斷:根據上述臨床癥狀和基因突變的結果,該患兒診斷為ARO,TCIRG1基因外顯子3雜合移碼突變 (3705C/-),根據人類基因突變數據庫 (http://www.hgmd.org/),此突變為新發現的ARO致病突變位點。
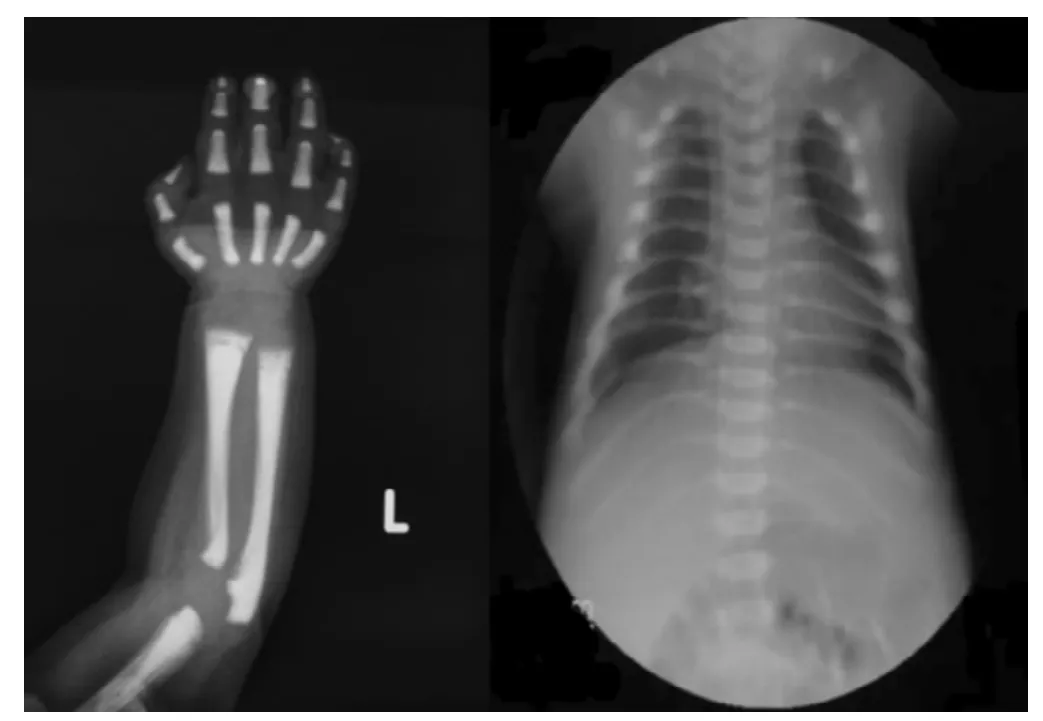
圖1 ARO患兒左上肢和胸部X線檢查所示Figure 1 X-ray radiography of the ARO infant's left upper limb and chest
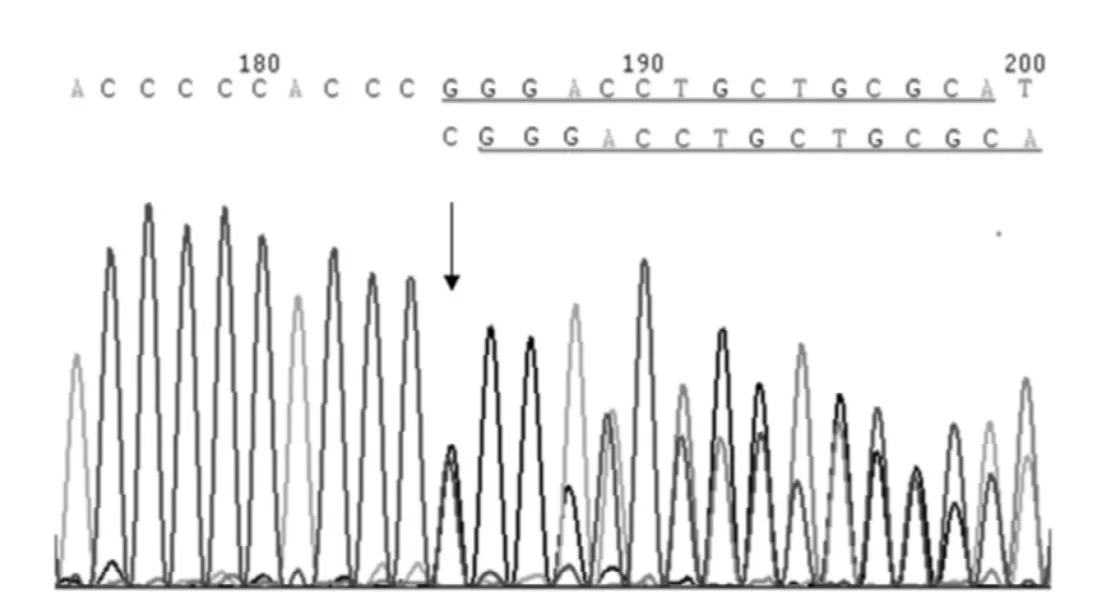
圖2 TCIRG1基因3號外顯子78位C缺失發生雜合移碼突變(3075C/-,見箭頭所指)Figure 2 Genetic analysis showing the novel frameshift mutation in exon 3 of TCIRG1 gene
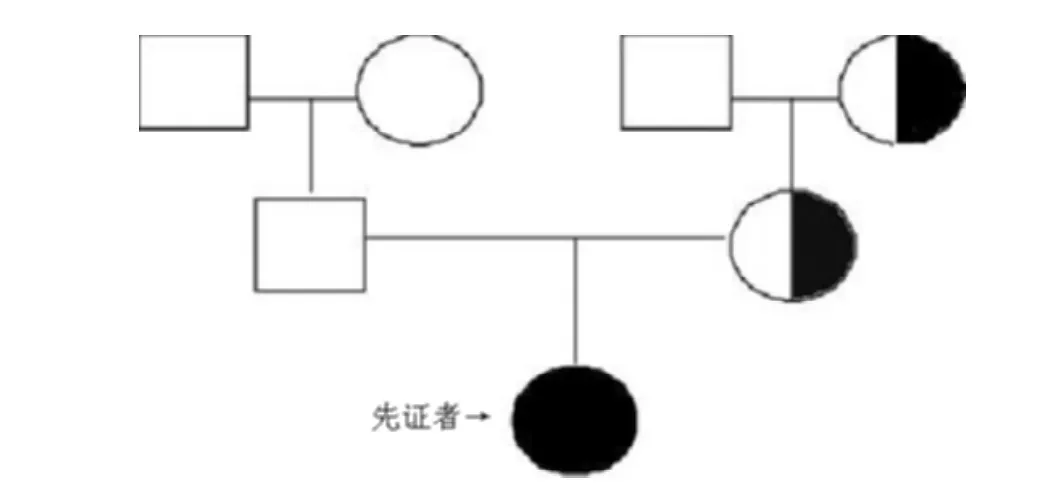
圖3 本例ARO患兒家系圖Figure 3 The pedigree of the ARO infant
2 討論
ARO是根據遺傳模式定義的骨硬化癥中一種比較少見的類型,是具有嚴重臨床表現的嬰兒期常染色體隱性骨骼疾病。臨床表現為再生障礙性貧血、血小板減少癥、肝脾腫大、神經精神運動障礙和危及生命的骨髓衰竭。X線檢查可以發現全身性的骨骼普遍硬化。TCIRG1、CLCN7和OSTM1基因是ARO的3個主要致病基因[2-5]。本文報道的患兒在6月齡時根據嚴重貧血、血小板減少、肝脾腫大、典型高骨密度X線表現和TCIRG1基因3號外顯子78位核苷酸雜合移碼突變 (3705C/-)診斷為ARO。患兒的母親和外祖母也是此基因突變的攜帶者,但未發病,而其父親為正常野生型基因。ARO是常染色體隱性遺傳性骨硬化癥,患者應該為該突變位點的純合子才會發病,而在本研究中,患兒為該突變位點的雜合子。通過復習文獻,有研究發現部分ARO患兒雖然是TCIRG1基因單等位基因突變的攜帶者,卻以類似于常染色體顯性遺傳的方式發病[5,8-9]。對此該研究指出 Alu-介導的同源重組和雜合子水平的同一基因組大片段缺失可能是ARO患者在單等位基因突變的情況下發病的潛在機制,但這些變化是目前常規的突變檢測方法所無法發現的[5]。本研究已經對患兒及其母親進行了Alu-介導的同源重組的研究,但未發現相關證據,故推測此TCIRG1基因是該ARO患兒的致病基因,其3號外顯子雜合移碼突變 (3705C/-)是新發現的與ARO有關的突變位點,需要進一步的研究明確患兒攜帶單等位基因突變的情況下發病的機制。
本研究中ARO患兒的ALP水平高于同年齡正常參考值。由于兒童處于生長發育期,血清ALP水平可作為反映成骨細胞活性的骨轉換指標,兒童的正常參考值高于成年人。通常骨硬化癥被認為是一種由于破骨細胞數量或者功能缺陷導致的骨吸收障礙疾病。但是以往亦有文獻報道ARO患者血清ALP水平升高[10-11],此現象顯示與其他類型的骨硬化癥患者相比,ARO患者的成骨細胞分化和功能可能存在差異。而在ARO患兒骨活檢中也證實發現了活躍的成骨細胞[10]。在CLCN7基因敲除小鼠中血清ALP水平上升30%也提示骨形成和骨吸收的平衡被打破[12]。近年來一些研究也證實在骨硬化癥發病機制中成骨細胞可能起了一定的作用[13-15]。因此,增高的血清ALP水平可能反映了在年幼的ARO患者中成骨細胞和破骨細胞之間平衡的調節。進一步的研究需要著眼于揭示骨硬化癥中成骨細胞-破骨細胞相互關系背后的病理生理機制。
綜上所述,TCIRG1基因突變是中國ARO患者的致病基因之一,此基因突變可導致肝脾腫大、嚴重貧血和血小板減少癥等嚴重的臨床表現,因此基因檢測對于診斷骨硬化癥至關重要。進一步的功能研究有助于揭示此ARO患者血清ALP增高和攜帶單等位基因突變即發病的可能的機制。
1 Stark Z,Savarirayan R.Osteopetrosis [J].Orphanet J Rare Dis,2009,4:5.
2 Bliznetz e A,Tverskaya SM,Zinchenko RA,et al.Genetic analysis of autosomal recessive osteopetrosis in Chuvashiya:the unique splice site mutation in TCIRG1 gene spread by the founder effect[J].Eur J Hum Genet,2009,17(5):664 -672.
3 Phadke SR,Fischer B,Gupta N,et al.Novel mutations in Indian patients with autosomal recessive infantile malignant osteopetrosis[J].Indian J Med Res,2010,131:508 -514.
4 Besbas N,Draaken M,Ludwig M,et al.A novel CLCN7 mutation resulting in a most severe form of autosomal recessive osteopetrosis[J].Eur J Pediatr,2009,168(12):1449 -1454.
5 Pangrazio A,Caldana ME,Sobacchi C,et al.Characterization of a novel Alu-Alu recombination-mediated genomic deletion in the TCIRG1 gene in five osteopetrotic patients [J].J Bone Miner Res,2009,24(1):162-167.
6 Lam CW,Tong SF,Wong K,et al.DNA -based diagnosis of malignant osteopetrosis by whole-genome scan using a single-nucleotide polymorphism microarray:standardization of molecular investigations of genetic diseases due to consanguinity[J].J Hum Genet,2007,52(1):98-101.
7 Zhang ZL,He JW,Zhang H,et al.Identification of the CLCN7 gene mutations in two Chinese families with autosomal dominant osteopetrosis(typeⅡ)[J].J Bone Miner Metab,2009,27(4):444-451.
8 Sobacchi C,Frattini A,Orchard P,et al.The mutational spectrum of human malignant autosomal recessive osteopetrosis[J].Hum Mol Genet,2001,10(17):1767.
9 Kornak U,Schulz A,Friedrich W,et al.Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause infantile malignant osteopetrosis[J].Hum Mol Genet,2000,9(13):2059.
10 Taranta A,Migliaccio S,Recchia I,et al.Genotype-phenotype relationship in human ATP6i-dependent autosomal recessive osteopetrosis[J].Am J Pathol,2003,162(1):57 -68.
11 Delfattore A,Peruzzi B,Rucci N,et al.Clinical,genetic,and cellular analysis of 49 osteopetrotic patients:implications for diagnosis and treatment[J].J Med Genet,2006,43(4):315 -325.
12 Neutzsky-wulff AV,Karsdal MA,Henriksen K.Characterization of the bone phenotype in ClC -7-deficient mice[J].Calcif Tissue Int,2008,83(6):425-437.
13 De Vernejoul MC,Kornak U.Heritable sclerosing bone disorders:presentation and new molecular mechanisms[J].Ann N Y Acad Sci,2010,1192(1):269-277.
14 Marzia M,Sims NA,Voit S,et al.Decreased c-Src expression enhances osteoblast differentiation and bone formation [J].J Cell Biol,2000,151(2):311-20.
15 Del Fattore A,Cappariello A,Teti A.Genetics,pathogenesis and complications of osteopetrosis[J].Bone,2008,42(1):19-29.
The Clinical Phenotypes and Family Study of Autosomal Recessive Osteopetrosis Caused by TCIRG 1 Mutation
WANG Chun,HE Jin -wei,FU Wen -zhen,et al.Metabolic Bone Disease and Genetics Research Unit,Department of Osteoporosis and Bone Diseases,Shanghai Sixth People's Hospital Affiliated Shanghai Jiaotong University,Shanghai 200233,China
ObjectiveTo study the clinical manifestations and molecular defect in an infant with autosomal recessive osteopetrosis(ARO)to improve the clinicians'understanding of this rare and malignant bone disease.Methods The biochemical parameters,abdominal ultrasound and X - ray were examined on this ARO infant.The entire coding region and adjacent splice sites of the T - cell immune regulator 1(TCIRG1)gene were amplified and sequenced directly in the patient,her parents,her paternal grandparents and her maternal grandparents.One hundred healthy donors were recruited as controls for mutation analysis.ResultsThe infant had severe anemia,thrombopenia and hepatosplenomegaly.Her hemoglobin was only 74 g/L.Her serum ALP level was 1 115 U/L.X -ray images of the chest and upper limb showed typical high bone density.A novel heterozygous frameshift mutation in exon 3 of the TCIRG1 gene(3705C/-)was detected in the infant,her mother and maternal grandmother.No controls harbored mutations in the TCIRG1 gene.ConclusionThe newly-discovered heterozygous mutation in exon 3 of the TCIRG1 gene was responsible for the Chinese infant with ARO,which clinical manifestations are severe anemia,thrombopenia and splenomegaly.So Genetic Analysis is necessary to diagnose osteopetrosis.The following functional study will investigate the potential mechanism to explain the elevated level of serum ALP and the existence of ARO in patient with monoallelic mutation.
Osteopetrosis;Genetic diseases,inborn;T -cell immune regulator 1 gene
R 681.4
A
1007-9572(2011)12-4070-03
上海市科委重大科技專題攻關專項 (10DZ1950100),上海市科委上海市自然科學基金 (11ZR1427300),上海市科委攻關-生物醫藥 (08411963100),國家自然科學基金面上項目 (81070692),國家自然科學基金青年項目 (81000360)
200233上海市,上海交通大學附屬第六人民醫院骨質疏松和骨病專科,骨代謝病和遺傳研究室
章振林,200233上海市,上海交通大學附屬第六人民醫院骨質疏松和骨病專科,骨代謝病和遺傳研究室;E-mail:zzl2002@medmail.com.cn
2011-09-05;
2011-11-03)
(本文編輯:趙躍翠)
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
