硒的伏安行為及測定的研究進展
2011-06-12 06:31:14王瑞俠周享春陸光漢
武漢工程大學學報 2011年12期
王瑞俠,周享春,陸 蓉,陸光漢
(1.池州學院化工系,安徽 池州 347000;2.長江大學化學環境工程系,湖北 荊州 434023;3.武漢職業技術學院旅游系,湖北 武漢 430074;4.武漢華中師范大學化學系,湖北 武漢 430079)
0 引 言
硒是人體必需要的微量元素之一,具有預防和抑制腫瘤的作用.硒為電子及冶金工業領域所使用,所以硒的分析顯得特別重要.硒和碲都是多價元素,對于碲的電化學行為筆者已作綜述[1-2].由于硒變價的多樣性,導致電化學反應的復雜性.它們表現出特殊的電化學性質.硒可以與某些金屬生成金屬互化物,也可以生成硒的絡合物,還可以生成H2Se等氫化物,溶出伏安法[3]就是利用這些特性建立起硒的電化學分析法.筆者在此對硒的陽極和陰極溶出伏安法的行為作出評述.
1 硒同金屬離子形成互化物的伏安行為
用伏安法(懸汞電極作工作電極)測定Se(IV)[4]時,一般是先在一定電位下富集,然后溶出,產生溶出電流.電極反應式為:
H2SeO3+4H++Hg+4e→HgSe+3H2O
HgSe+2H++2e→H2Se+Hg
文獻[5]用玻碳汞膜電極作為工作電極,在0.1 mol/L HClO4-0.1 mol/L KCl介質中,Se(IV)在-0.1 v(vs·SCE)電解,硒形成難溶的HgSe,陰極溶出時,富集在電極上的HgSe還原電極反應如下:
H2SeO3+6H++6eH2Se+3H2O
Hg+H2SeHgSe+2H++2e
HgSe+2H++2eH2Se+Hg
其他工作見文獻[6-15]和表1.
為了提高測定靈敏度,利用硒與其他金屬離子生成金屬互化物的性質,在測定體系中引入金屬離子(如Pb2+,Cu2+,Hg2+等),如在測定體系中加入Cu2+[16],Cu2+與Se(IV)生成金屬互化物,反應如下:
Se(IV)+2Cu(Hg)+4e→Cu2Se(Hg)2
然后再溶出:
Cu2Se(Hg)2+2H++2e→H2Se+2Cu(Hg)
這種方法靈敏度大大提高.文獻[16]的檢測限達9×10-10mol/L.除了加上述金屬離子外,還可以加入稀有金屬離子,Wang[25]在0.1 mol/L H2SO4-10 μg/L Rh(Ⅲ)體系中,在-0.2 V集時,發生下述電極反應:
3H2SeO3+12H++2Rh(Ⅲ)+18e→Rh2Se3+9H2O
產生的伏安響應在-0.97 V檢測限達6×10-12mol/L.
在0.3 mol/L HCl-75PPbRh(Ⅲ)體系中,由于變價硒的復雜性,Se(IV)的伏安行為與Wang不一樣.硒與介質的組分生成混合絡合物,并產生氫催化波[17].Se(IV)于-0.2 v(vs·Ag/AgCl)沉積,Se(IV)被還原成Se(-Ⅱ),接著Se(-Ⅱ)與Rh(Ⅲ)等生成混合絡合物:

當電位掃描-1.15 V(vs·Ag/AgCl),發生下面電極反應:
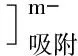

表1 硒的伏安行為
產生的電流是催化氫波引起的,產生催化氫波的原因是混合絡合物吸附在汞電極表面,使電極表面得到修飾,H+放電電位正移.又因為下列反應發生:
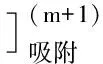
產物又回到電極上放電,形成催化循環而增大電流,產生靈敏的催化氫波,檢測限達2.4×10-12mol/L.用銅汞齊[18]作工作電極,測定硒時發生下述電極反應:
沉積:Cu2++2e+Hg→Cu(Hg)
2Cu(Hg)+H2SeO3+4H++4e→Cu2Se(Hg)2
溶出:Se(Hg)2+2H++2e→H2Se+2Cu(Hg)
檢測限達0.25 nmol/L.
文獻[19]考慮到汞害和環境污染的問題,用碳金膜電極微分陽極溶出伏安測定水中痕量Se(Ⅳ)和Se(Ⅵ),在0.5 mol/L HNO3底液中,于-0.4 V(vs·SCE)電積,然后溶出,其過程如下:
該方法檢測限達5×10-10mol/L.
其他工作見表2[26-33].
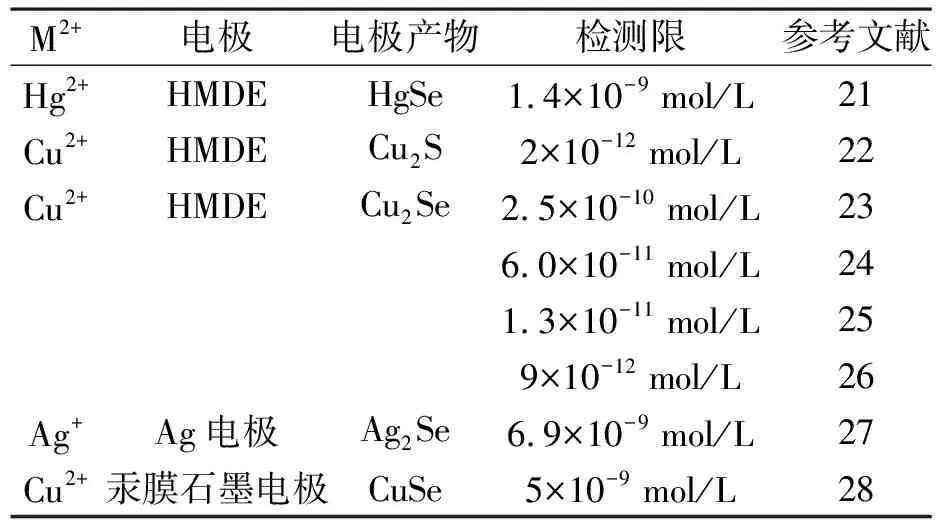
表2 在不同物質存在下硒的陰極溶出伏安測定
2 硒絡合物的伏安行為
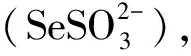
如文獻[20]研究了在R-B緩沖溶液中,硒(Ⅳ)與鄰苯二胺(O-PDA)體系的示差脈沖吸附伏安行為.Se(Ⅳ)在溶液中先與O-PDA生成絡合物,該絡合物吸附在汞電極上,然后溶出,其電極反應式如下:
Se(Ⅳ)-O-PDA(ads)(Hg)+4e→Se(O)-O-PDA(ads)(Hg)
該方法靈敏度高,檢測限可達5.0×10-10mol/L,用此法測定了人發中微量硒.
文獻[21]在0.1 mol/L HCl-1.0×10-3二氨基萘(DAN)體系中,Se(Ⅳ)-DAN絡合物在+0.05 V(vs·SCE)富集,Se(Ⅳ)+DAN→[Se(Ⅳ)-DAN]吸附
在-0.06 V(vs·SCE)Se(Ⅳ)還原成Se(0),與汞生成金屬互化物.
[Se(Ⅳ)-DAN]+4e+Hg→SeHg+DAN吸附
在-0.46 V(vs·SCE)進一步還原成Se(-II).
SeHg+2H++2e→H2Se+Hg
該方法的檢測限達到1×10-8mol/L.
文獻[22]報道了在0.1 mol/L HCl-4-苯二胺(4N0PD)溶液中,Se(III)與4N0PD發生反應.
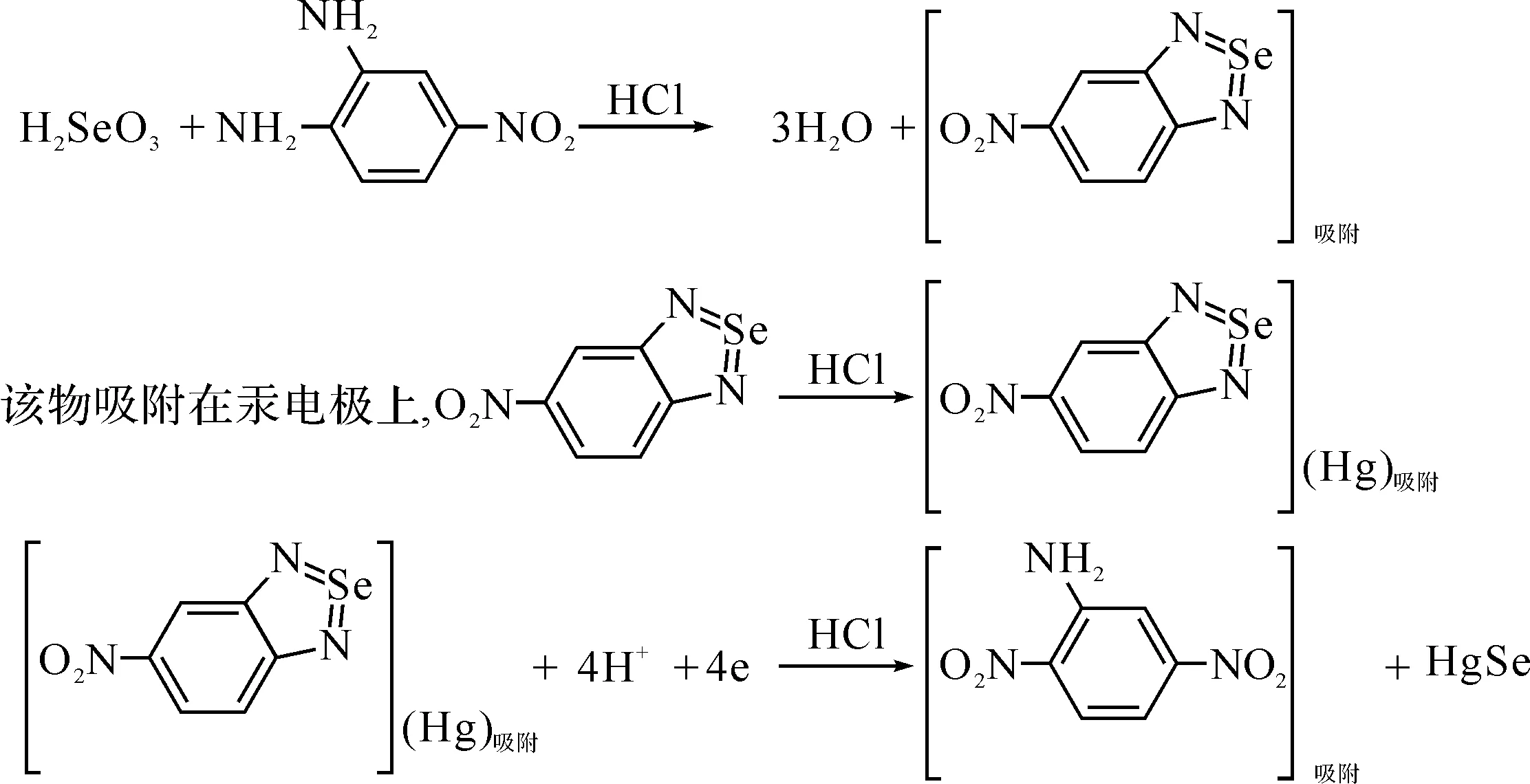
汞電極上HgSe進一步還原為:
HgSe+2H++2e→H2Se+Hg
該方法的檢測限7.6×10-10mol/L.
文獻[23]利用3,5-二溴代鄰苯二胺在弱酸(0.1 mol/L HNO3)介質中與硒反應生成4,6-二溴代苯硒胺,該絡合物有電活性,電極反應機理為:
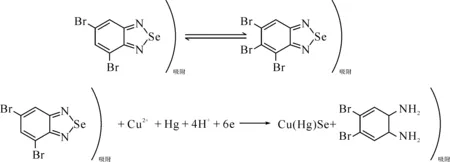
得出:

由于汞(膜)電極對人體有害,而鍍金膜電極要耗掉貴重的黃金.
而鉍膜玻碳電極電位窗口寬,溶出峰分辯力強,對溶解氧不敏感,實驗時不必除O2,方便了實驗操作.而且鉍膜電極是對環境友好的電極,鉍膜電極的使用,使溶出伏安的技術得到改進和發展,近幾年各種修飾電極在溶出伏安法中的應用[33-35],使得該方法具有更多的實際應用價值和良好的發展空間.文獻[24]采用鉍膜玻碳電極作為工作電極在HAC-NaAc~氨基苯(ABSA)(pH2.9)的體系中測定硒.測定的原理是Se(Ⅳ)與ABSA生成絡合物,由于該絡合物具有強烈的吸附性,吸附在鉍膜電極上的絡合物進行電還原,產生溶出電流.檢測限達1.3×10-8mol/L.
電極反應機理如下:
2ABSA+H3SeO3+2H+→ABSA-Se-ABSA+3H2O
Se(Ⅳ)(ABSA)2+Bi→Se(Ⅳ)(ABSA)2(ads)(Bi)
Se(Ⅳ)(ABSA)2(ads)(Bi)+4e→Se(0)(ABSA)2(ads)(Bi)
文獻利用在0.5 mol/L HCl-0.5 mol/L Kl溶液中,Se(Ⅳ)與I-作用生成Se-I2絡合物[37],該絡合物富集在汞電極上,吸附在汞電極上的Se(0)被還原成Se(-II)產生電流.實際上Se-I2絡合物1951年就有作者把它用來測定海水中硒(光度法),但用在溶出伏安法測定微量硒的確是一種創新.
其他生成硒絡合物的溶出伏安法見文獻[38-40].
文獻[41]用鉍膜石墨電極測定硒時,產生一催化氫波,電位為1 150 mV(vs·Ag/AgCl),并被實驗證明的確為一催化氫波;作者用同樣的催化氫波體系測定了另一元素[42].
還有方法把陰極溶出伏安與催化極譜聯用,使測定靈敏度大為提高.它的原理是在一定的電位下富集,然后把電極放入有催化體系的溶液中,電極反應如下:
電沉積:Hg+H2SeO3+4H++4e→HgSe+3H2O
溶出:HgSe+2H++2e→Hg+H2Se
生成的H2SeO3又在電極上還原,產生很大的催化電流,靈敏度達到7.0×10-10mol/L.
為了提高靈敏度,有方法先把硒富集在一個載體上[43-44],然后進行收集,再用伏安法測定.實踐證明,用伏安法測定硒是一簡單可行的方法,特別在研究其反應機理方面更有特色.
3 結 語
由于溶出伏安法可以有效地提高靈敏度,降低檢測限,溶出伏安儀價廉,它是目前乃至今后相當長時間內測定微量硒的主要方法之一.但任何一種分析方法都有不足,溶出伏安法存在最大的難題是工作電極的表面(特別是固體電極)的污染,影響了電極的穩定性.隨著使用方便的電化學敏感電極深入研發,前處理樣品方法聯用、清洗和再生電極表面的技術不斷改進,測定硒體系不斷的更新以及電極反應機理的深入研究,必將使伏安法技術得到新的發展.
參考文獻:
[1]陸光漢,黃渝卿.碲的極譜分析概況[J].冶金分析,1990,10(3):29-32.
[2]陸光漢,何治柯,劉傳銀.溶出伏安法測定碲的概況[J].冶金分析,1992,12(4):37-39.
[3]Achterberg E P,Barriacla J L,Braungardt C B.Cathodic stripping voltammetry[J].Encycloped of analytical science,2005(2):203-211.
[4]Danielc S.Anodic stripping voltammetry[J].Encycloped of analytical science,2005(2):197-202.
[5]鄒家慶,羅平,宋軍,等.1.5次微分陰極溶出伏安法測定水樣中痕量硒[J].南京化工大學學報,2001,23(4):50-53.
[6]徐暉,張必成,王升富.微分脈沖陰極溶出伏安法測定環境水樣中的痕量硒[J].環境化學,2001,20(4):386-391.
[7]Elleouei C,Queniel F,Madec C.Determination of inorganic and organic selenium species in natural by cathodic stripping voltammetry[J].water Research,1996,30(4):909-914.
[8]Ochsenkuhn-Perropoulou M,Tsopelas F.Speciation analysis of selenium suing voltammetric techniques[J].Anal Chim Acta,2002,467(1/2):167-178.
[9]Recai I,Guler S.Determination of selenium in garlic by cathodic stripping voltammetry[J].Food Chemistry,1999,66:381-385.
[10]Recai I,Guler S.A direct method for the determination of selenium and lead in cow's milk by differential pulse stripping voltammetry[J].Food Chemistry,2000:345-350.
[11]Anca-Iulia S,Gobriela-Raluca B,Emilia-Elena L,et al.Differential pulse cathodic stripping voltammetric determination of selenium in pharmacentical products[J].Journal of Phormacentical and Biomedical of Analysis,2002,30(4):1425-1429.
[12]Zvonimir S,Jaroslava S G,Nikala M,et al.Development of a Chronopotentionetric stripping method for the determination of selenium(Ⅳ) in mixed diets[J].Food Chemistry,2005,92:771-776.
[13]張曉麗,王麗增,馬成松,等.痕量硒的伏安法測定[J].山東大學學報,1995,30(2):186-189.
[14]Suznjevic D,Blagojevic S.Determination of selenium CSV suing copper microelectrode[J].Microchem J,1997,57:255-260.
[15]藏樹良,王歆睿,鐵梅,等.陰極溶出伏安法測定痕量硒[J].遼寧大學學報:自然科學版,2005,32(4):289-292.
[16]Monica P,Luigi F,Patrizia M,et al.Determination of selenium in Italian rices by differential pulse cathodic stripping voltammetry[J].Food Chemistry,2007,105:1091-1098.
[17]Britta L,Constant M G,Van D B.Detemination of selemium by catalytic cathodic stripping voltammetry[J].Anal Chim Acta,2000,418:33-42.
[18]Robert P,Wlodystaw W K.Determination of trace selenium on hanging copper amalgam drop electrode[J].Electrochimica Acta,2007,53:584-589.
[19]李發生,王素芳.玻碳金膜電極線性掃描微分陽極溶出伏安法分析測定水中痕量Se(Ⅳ)和Se(Ⅲ)[J].中國環境監測,1994,10(2):44-48.
[20]孫長林,王建燕,李學斌,等.硒的汞膜電極示差脈沖吸附伏安法[J].分析化學,1991,19(2):139-142.
[21]Tanaka S H,Sugawara K,Taga M.Voltammetry of selenium based on an adsorptive accumulation of selenium-2,3-diamino naphthalene[J].Anal Sci,1990(6):475-480.
[22]Ashournia M.Determination of Se(Ⅳ) in natural by adsorptive stripping voltammetry of 5-nitropiazselenol[J].Journal of Hazardous Materials,2010,174:788-794.
[23]蔡乾濤,王永麗,陸曉華.吸附溶出伏安法測定微量硒-銅離子對二溴代苤硒腦體系增敏作用[J].分析化學,1991,19(1):27-31.
[24]Zhang Qing,Li Xiang Jun,Shi Hui,et al.Determination of trace selenium by differential pulse adsorptive stripping voltammetry at a bismuth film electrode[J].Electrochimica Acta,2010,55:4717-4721.
[25]Wang J,Lu J.Uitratrace measurements of selenium by cathodic stripping voltammetry[J].Anal Chim Acta,1993,274:219-224.
[26]Muhamed R,Wel L.Analysis of selenium species using cathodic stripping voltammetry[J].J Technol,2006,44:55-66.
[27]Fommaso F,Silvia R,Puola S.Simultaneous determination of the speciation of selenium and tellurium ingeological matrices by use of on iron(III)-modified chelating resin and cathodic stripping voltammetry[J].Anal Chim Acta,1998,361:113-123.
[28]Korolczuk M,crabarczyk M.Determination of Se(Ⅳ) in on-line system by cathodic stripping voltammetry[J].Electroanalysis,2003,15:821-826.
[29]Van C M G,Berg D,Khan S H.Determination of selemium in sea water by cathodic stripping voltammetry[J].Anal Chim Acta,1990,231:221-229.
[30]Lenge B.Determination of trace selenium by cathodic stripping voltammetry[J].Anal Chim Acta,2000,418:33-37.
[31]Papoff T,Boeci F,Lanca N F.Speciation of selenium in natural water and snow by DPCSV at the hanging mercury Drop electrode[J].Microchem J,1998,59:50-76.
[32]Recai I,Giiler S,Banu K.Determination of cadmium,Lead and selenium in medicago sativa herb by differential pulse stripping voltammetry[J].Anal sci,1999,15(5):493-495.
[33]Hasan A Y,Din G S.Stripping voltammetry of selenium(Ⅳ) in the presense of copper ion[J].Anal Sci,1989(5):89-92.
[34]Lu Guang Han,Wang Ling Yan,Song Feng,et al.Determination of uric acid and Norepinephrine by chitosan-multiwall carbon nanotube modified Electrode[J].Electroanalysis,2005,17(10):901-905.
[35]Lu Guang Han,Yao Xin,Wu Xiao Gang,et al.Determination of the total iron by chitosan-modified glassy carbon electrode[J].Microchem J,2001,69:81-87.
[36]Lu Guang Han,Yao Xin,Zhou Xiang Chun,et al.Determination of trace thiocyanate by a chitosan-modifed glassy carbon electrode[J]. Chem J Chinese University,2002,18(3):316-320.
[37]Ashournia M,Aliakbar.Determination of selenium in natural water by adsorptive differential pulse cathodic stripping votammetry[J].Journal of Hazardous Materials,2009,168:542-547.
[38]Inam R,Somer C.Adsorptive stripping voltammetry of selenium(Ⅳ) in the thiocyanate suing ascorbic acid as the reductant[J].Anal Sci,1997,13:653-656.
[39]Stara V,Kapanica M.Cathodic stripping voltammetry and adsorptive stripping voltammetry of selenium [J].Anal Chim Acta,1988:231-236.
[40]Long J,Nagaosa Y.Determintion of selenium(Ⅳ) by catalytic stripping voltammetry with an in situ plated bimuth-film electrode[J].Anal Sci,2007,23:1343-1346.
[41]Long J J,Nagaosa Y.Cathodic stripping voltammetry determination of As(III) with in situ plated bismuth-film electrode using the catalytic bydrogen wave[J].Anal Chim Acta,2007,593:1-6.
[42]Bertolino F A,Jtorriero A A,Salinas R O R.et al.Speciation analysis of Se(Ⅳ) in natural water using square-wave voltammetry after preconcentration on activated carbon[J].Anal Chim Acta,2006,572:32-38.
[43]王勝天,許宏鼎,李景虹.環境電分析化學[J].分析化學,2002,30(8):1005-1011.
[44]Claudete F P,Gonzage F B,Santos A M G,et al.Determination of selenium by anodic stripping voltammetry using gold electrode made from recordable CDs[J].Talanta,2006,69(4):877-881.
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
現代企業(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
