氣相色譜-離子阱質譜聯用測定蔥中有機氯農藥的含量
2011-01-30 02:15:50龐小剛段迎燕劉文靜
質譜學報 2011年3期
張 洪,雷 昊,龐小剛,段迎燕,劉文靜
(山西省分析測試中心,山西太原 030006)
復雜基質中農藥殘留一直是農藥多殘留分析的難題。多級質譜技術有較強的選擇性,能為解決復雜基質中的干擾提供條件。隨著離子阱質譜的發展,利用其時間上的串聯特性,使得串聯質譜技術在農藥殘留分析中得到廣泛應用[1]。
目前,有機氯農藥的檢測一般采用氣相色譜法或氣相色譜-質譜法中的選擇離子掃描進行測定[2-4],但上述方法容易被基質干擾,造成結果假陽性。氣相色譜-離子阱串聯質譜法具有靈敏度高、檢出限低、確證性強、前處理相對簡單的特點,本工作采用該方法測定蔥中有機氯農藥的殘留[5]。
1 試驗部分
1.1 主要儀器與試劑
Thermo PolarisQ氣相色譜-質譜聯用儀:美國 Thermo公司產品,外置離子源;TGL-16M離心機:長沙湘智離心機儀器有限公司產品;T25分散器:德國 IKA公司產品;Envi-18柱:美國Supelco公司產品。
有機氯農藥混合標準品 (α-BHC、β-BHC、γ-BHC、δ-BHC、p,p′-DDE、p,p′-DDD、o,p′-DDE、p,p′-DDT):購自國家標準物質中心 ;乙腈(色譜級);氯化鈉(分析純);載氣:He(純度為99.999%)。
1.2 分析方法
1.2.1 提取 稱取20 g樣品于80 mL離心管中,加入40 mL乙腈,用高速組織搗碎機以15 000 r/min勻漿1 min,加入5 g氯化鈉,繼續勻漿1 min,將離心管放入離心機中,以3 000 r/min離心5 min,取上清液20 mL,待凈化。
1.2.2 凈化 先用10 mL乙腈預淋 Envi-18柱,再轉移20 mL提取液至小柱內,用15 mL乙腈洗脫并收集,40℃水浴中旋轉濃縮,氮氣吹干,定容至1 mL。
1.2.3 測定 PolarisQ外置離子源離子阱質譜,TR-5MS色譜柱;程序升溫:初始溫度60℃,保持1 min,以10 ℃/min升至 250 ℃,保持 8 min;進樣口溫度260 ℃;進樣量1μL;進樣方式:不分流進樣;載氣:恒流模式流速 1 mL/min;電離源:電子轟擊模式,70 eV;離子源溫度220℃;傳輸線溫度260℃。
樣品中農藥的質量色譜圖示于圖1。
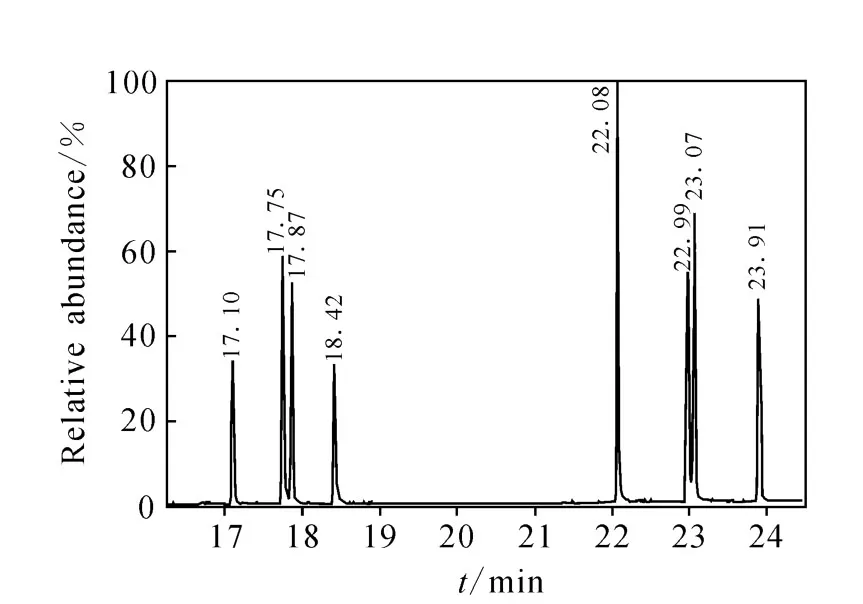
圖1 樣品中農藥的質量色譜圖Fig.1 The mass chromatograms of samples analyzed obtaind with GC-MS/MS
二級質譜的方法優化:盡量選擇質量數較大,相對豐度較高的離子作為母離子,這樣可以保證方法的靈敏度。母離子及碰撞誘導解離的電壓是分析的關鍵,能量不足,無子離子生成,能量過大,則產生過多的子離子,導致成分復雜,母離子完全消失。最佳二級質譜圖應該有兩個相對峰豐度大于50%的子離子,并有5%~10%的子離子。各農藥的二級質譜參數列于表1。
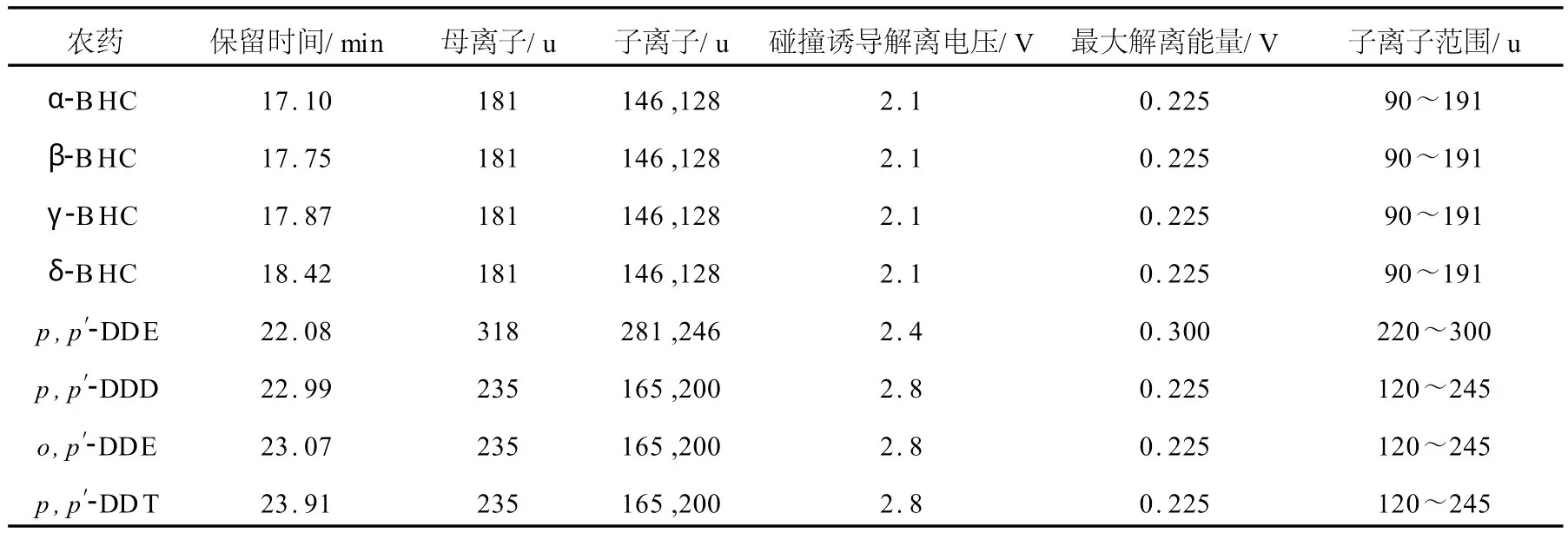
表1 各農藥的二級質譜參數Table 1 MS/MS parameters of pesticides
1.2.4 標準曲線的制作 將購買的農藥標準品稀釋成 5 個濃度梯度 (0.05、0.1、0.5、1、5 mg/L)的樣品,分別進樣,以峰面積和進樣濃度做標準曲線,其相關參數列于表2。
1.2.5 添加回收率實驗 分別準確添加0.05、0.5、5 mg/kg有機氯農藥的標準溶液于空白大蔥中,每個濃度設5個重復,其結果列于表3。

表2 農藥標準曲線參數Table 2 Carlibration parameters of pesticides
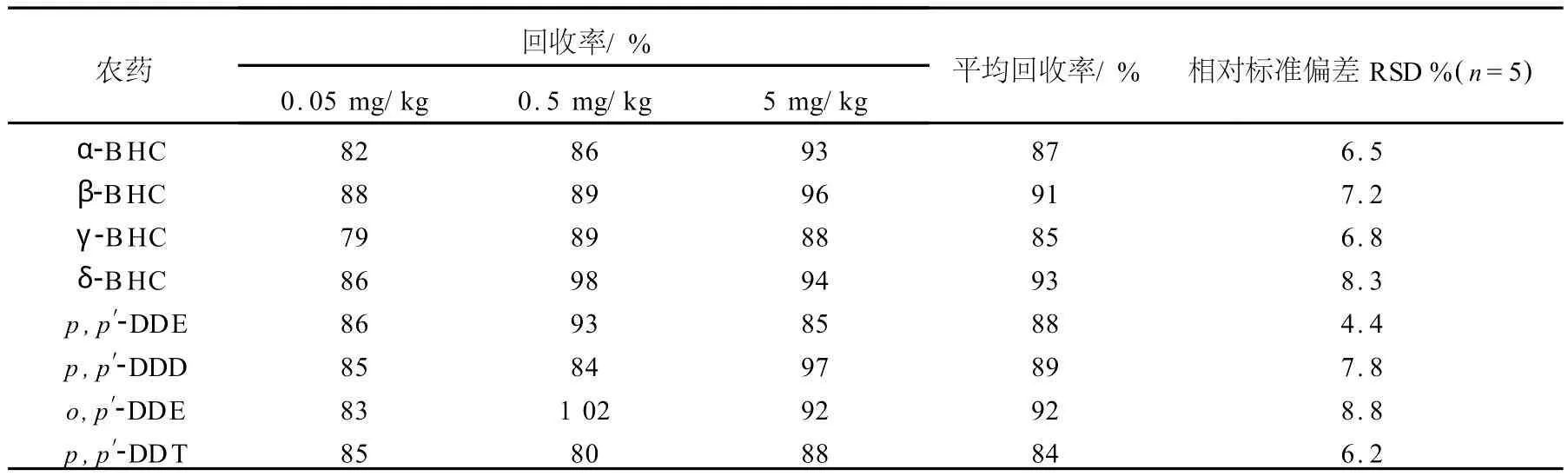
表3 農藥回收率實驗Table 3 Recoveries of pesticides
2 結果與討論
本實驗針對基質較為復雜的蔥作為本底,進行了8種有機氯農藥殘留分析。實驗結果表明,各種農藥的回收率均大于80%,相對標準偏差小于10%,線性相關良好,且最低檢出限為0.05 mg/kg,符合分析要求。由于每種農藥的碎裂都有其特定的規律性,所以根據串聯質譜所產生的二級離子,降低了基質干擾,簡化了前處理步驟,提高了方法的靈敏度,避免出現假陽性。
李重九等[6-8]利用Varian Saturn 2000氣相色譜-離子阱質譜聯用法進行農藥殘留檢測,但Varian Saturn 2000為內置離子源離子阱質譜,而本實驗使用的 Thermo PolarisQ為外置離子源離子阱質譜。相比較而言,內置離子源離子阱的離子化過程是在阱中完成,碎片離子相互撞擊的概率較大,因而會產生一些與裂解規律質量不相符的附加碎片離子;而外置離子源的離子化在阱外進行,減少了二級碎片離子的碰撞,產生附加較少的碎片離子,所以兩種儀器產生的離子碎片不盡相同,母離子和二級碎片離子的選擇也不完全相同,并且由于儀器的硬件配置不同,在產生二級碎片離子的過程中,所加的碰撞誘導解離的電壓也有差異。
[1]萬鄭凱,何 娟,康長安,等.氣相色譜質譜聯用在農藥殘留檢測方面的應用進展篇[J].分析測試技術與儀器,2006,12(1):51-58.
[2]任朝興.固相萃取-氣質聯用法同時測定海水中六六六滴滴涕狄氏劑[J].分析儀器,2009,13(5):59-61
[3]沈美芳,李 卿,杭蘇琴,等.水產品中六六六滴滴涕殘留檢測前處理方法的比較研究[J].食品科學,2006,27(7):191-195.
[4]黃建軍.測量大米中六六六滴滴涕殘留量不確定度評定 [J].廣西工學院學報,2005,16(3):23-26.
[5]中國疾病預防控制中心營養與食品安全所,農業部農藥檢定所.GB 2763—2005食品中農藥最大殘留限量[S].北京:中國標準出版社,2005.
[6]宋淑玲,李重九,馬曉東.蔬菜農藥多殘留分析中基質共提物凈化方法的研究[J].分析測試學報,2008,27(8):795-799.
[7]劉 珺,王大寧,李重九.林蛙油中農藥殘留的GC-MS/MS檢測方法[J].分析測試學報,2006,25(6):77-80.
[8]佟 玲,李重九.含硫蔬菜中50種農藥多殘留的氣相色譜串聯質譜檢測技術研究[J].分析測試學報,2008,27(9):930-935.
