蛋白質組學中質譜數據標準研究進展
2011-01-30 02:15:58馬海濱張紀陽孫漢昌謝紅衛
質譜學報 2011年3期
關鍵詞:標準
馬海濱,張紀陽,2,劉 輝,2,孫漢昌,2,謝紅衛
(1.國防科學技術大學,機電工程與自動化學院自動控制系,湖南長沙 410073;2.軍事醫學科學院,放射與輻射醫學研究所,北京蛋白質組研究中心,蛋白質組學國家重點實驗室,北京 102206)
在后基因組時代,蛋白質組學(proteomics)成為生命科學研究中的一個熱點[1]。由于缺乏類似基因組 PCR(polymerase chain reaction)擴增的樣品倍增方法,蛋白質組學研究對實驗技術的要求更高,因此,重復實驗、多實驗平臺互補、多策略互補等方法在蛋白質組學中更加重要。這些方法是提高蛋白質鑒定和定量結果覆蓋率及重復性的重要手段。目前,蛋白質組學中使用的主要研究策略大都基于質譜實驗與分析。質譜方法具有高通量和高靈敏度的特點,是蛋白質組學研究的一項支撐技術[2-3]。
由于質譜儀種類繁多,精度和性能差異較大,實驗產出數據格式多樣,實驗數據難以整合[4],而后續質譜數據處理的目的卻是要從實驗產出的海量數據中完成數據的獲取、處理、存儲和解釋。在目前的研究發展階段,實驗策略和數據分析方法種類繁多,而且還在不斷提出新的數據分析方法,在不同分析策略中使用的算法也不盡相同,在不同分析流程中使用的數據格式繁多且大多互不兼容,部分質譜數據文件格式列于表1。這些因素給數據共享和交換帶來困難,不利于分析結果的整合,與數據處理的目標背道而馳。因此,有必要對質譜數據處理中的數據格式問題進行研究。2004年以來,相繼提出了多種開放式數據標準并得到不同程度的應用,初步緩解了目前面臨的質譜數據格式兼容性的問題。
本工作對目前已有的質譜數據標準進行綜述,介紹質譜數據標準的研究現狀,比較各種數據標準的特點與優缺點,并展望質譜數據標準可能的發展方向。
1 質譜數據標準研究現狀
目前,蛋白質組學中質譜數據標準的主要制定組織是HUPO-PSI(Human Proteome Organization-Proteomics Standards Initiative)[5],此外系統生物學研究所(Institute for System Biology,ISB)[6]和歐洲生物信息學研究所(European Bioinformatics Institute,EBI)[7]也參與了質譜數據標準的制定。
2002年,在華盛頓召開的 HUPO會議上成立了PSI,其主要目標就是要在蛋白質組學領域中為數據表示定義公共數據標準,以解決蛋白質組學研究中數據格式不統一的問題,實現數據的比較、交換和驗證[8]。質譜數據公共標準和控制字(controlled vocabulary)的制定工作主要由PSI的MS(mass spectrometry)組織完成。由于可擴展標記語言[9](extensible markup language,XML)是一種與平臺無關的結構性信息表示方法,因此當時以及后來制定的數據標準主要是基于XML格式的[10]。數據標準不僅要支持多種質譜實驗技術,還必須能夠存儲與質譜實驗有關的MIAPE[11-12](the minimum information about a proteomics experiment)信息。
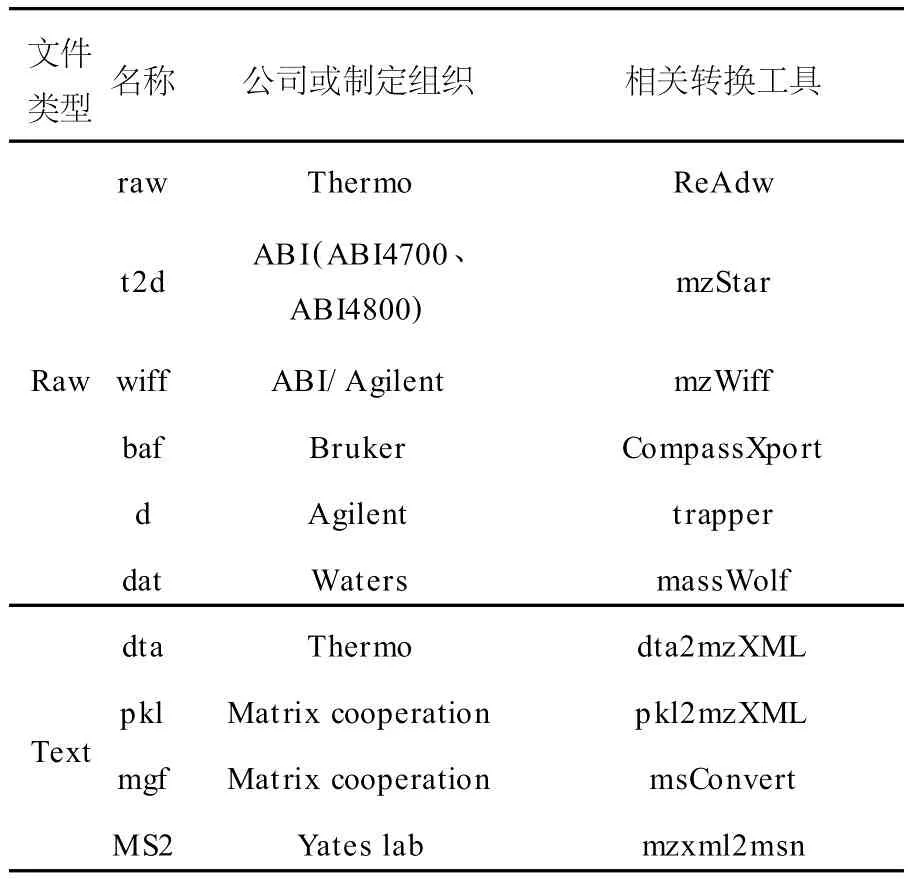
表1 質譜數據文件格式Table 1 File type of mass spectrometry data
2004年至2008年,用于存儲和交換原始數據的標準主要有mzData與mzXML。2008年,PSI主持發布了 mzML,試圖完全取代 mzData與mzXML。2003年,在蒙特利爾(Montreal)召開的 HUPO大會上,PSI-MS發布了 mzData 1.0版[13],目前使用的是2006年發布的mzData 1.05版。因其自身存在不足,該數據標準的應用相對比較有限。相對于mzData,ISB開發制定的數據標準 mzXML[14]得到了更廣泛的應用。而在SPC(seattle proteome center)開發的數據分析平臺 TPP(the trans-proteomic pipeline)中,使用的數據標準是 mzXML、pepXML和protXML,并試圖兼容 mzML。2009年,PSI將AnalysisXML[15]中蛋白質鑒定部分的數據標準更名為mzIdentML,作為一個獨立的數據標準發布,而關于蛋白質定量部分的數據標準暫時命名為mzQuantML[16]。另外,EBI在開發和維護數據庫PRIDE(PRoteomics IDEntifications database)時,為其中的數據提供了一個專門的數據標準 Pride XML,該格式將 mzData整合其中,作為其保存質譜數據的數據格式,其余部分實現對搜庫結果與實驗信息的存儲。表2列出了常用的數據標準及其制定組織,其中的數據分析階段參照TPP中的劃分。典型蛋白質鑒定流程示于圖1。
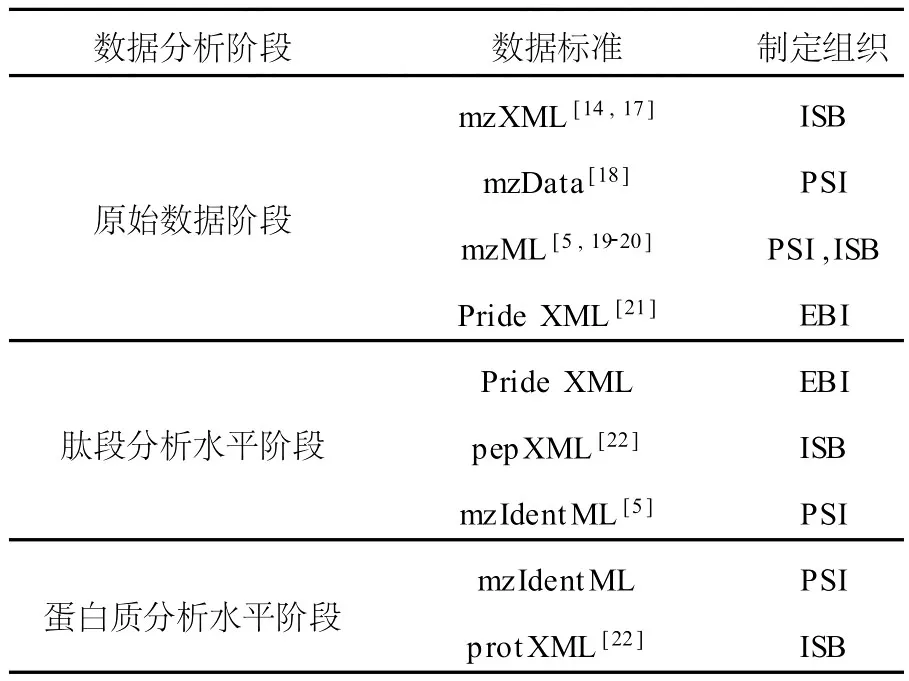
表2 現有質譜數據標準及其制定組織Table 2 Proteomics data format standards and corresponding organizations
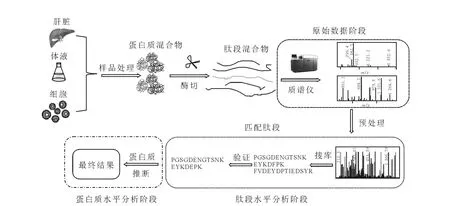
圖1 典型蛋白質鑒定流程Fig.1 Typical workflow of protein identification
2 質譜數據標準介紹
蛋白質組學研究的一個重要工作就是蛋白質鑒定。在從質譜實驗獲得質譜數據,直到獲得蛋白質鑒定結果的整個過程中,需要對不同格式的數據進行格式轉換和數據整合,以滿足不同數據分析軟件的輸入要求。在沒有制定統一的數據標準之前,不統一的數據格式極大的限制了蛋白質組學數據的共享和發表,制約了相關數據庫的開發,不利于研究人員對已有的研究成果進行再分析、整合和總結,導致不斷的重復鑒定。隨著研究工作的不斷深入和研究成果的不斷發表,構建蛋白質組學數據庫就顯得極為重要,而在整理數據時更需要一個統一的數據標準來對數據庫中的數據進行管理[14,23-25]。
下面介紹基于質譜技術的蛋白質組學中常用的數據標準。在 TPP中,將數據處理過程劃分為3個階段:原始數據階段、肽段水平分析階段和蛋白質水平分析階段[26]。本節將按照這3個階段分別介紹各種數據標準。
2.1 原始數據階段
該階段數據標準的主要作用是將質譜儀生產商的原始數據格式轉換為開放結構數據,以方便研究人員對數據的再次分析和對現有算法做出改進。這一階段目前使用的數據標準主要有mzData,mzXML和mzML。其中,mzML是針對原始數據階段最新制定的數據標準,它結合了前兩種標準的優點。
2.1.1 mzData數據格式的特點 mzData的最大特點是使用XML模型外的控制字來描述與設備和實驗設計等有關的參數。當采用新型儀器或新的實驗方案時,這些參數能夠以一個統一的數據格式存儲在數據文件中。控制字可分成控制字參數(cvParam)和用戶參數(userParam)兩種。控制字參數具有一定的固定性,用戶自定義的控制字可以放在用戶參數中。使用控制字既保證了數據格式的可擴展性,又保證了數據格式的靈活性。但是,由于沒有采取一定的機制限制用不同的方式編碼本質相同的信息,導致同一版本之間控制字的不一致,嚴重影響了數據的共享以及讀寫軟件的通用性。而且mzData中沒有使用索引,不能實現對數據文件中質譜圖的快速隨機讀取。
2.1.2 mzXML數據格式的特點 mzXML是用于存儲和交換質譜數據的開放數據格式,具有很高的靈活性,且能存放多種類型的數據——從未經任何處理的數據到經過深度數據處理的數據(如質心化、峰檢測等)。
在質譜實驗分析中,使用統一的數據標準可以方便地將新型質譜儀加入到數據分析平臺中,便于實驗數據的交換和發表,能為新的數據分析工具的開發提供統一平臺,因此mzXML從發布至今,已經得到廣泛的應用。但相對于二進制文件而言,基于XML的數據文件還存在一些不足,主要有兩點:一是將原始文件轉換成基于XML的數據文件,文件容量會增加。現在高精度質譜儀在1 h內的數據產出量會超過1 GB,數據經轉換后會帶來一定的存儲問題。而且,在XML文件中不能直接包含二進制數據,需要轉換成人工可閱讀的數據文件,這就不可避免地造成文件容量的增加;二是降低了數據文件中信息的讀取速度[14]。雖然在mzXML文件中使用了索引,避免了數據文件必須從開始讀到結尾的弊端,但還不能實現按條件對圖譜和數據信息進行讀取。分析mzXML文件結構還可以得出:該數據文件并不適合于計算,而且也沒有存儲與實驗設計有關的參數信息[17]。
2.1.3 mzML的發展進程 2008年以前,主要有兩大開放數據標準:mzData和 mzXML。兩種標準處理的是相同的數據信息,這勢必增加軟件開發人員的負擔。因此,mzData和mzXML的制定組織聯合儀器生產廠商、數據分析人員和一些終端用戶,在 HUPO-PSI的贊助下[27],開發了新的數據標準——mzML,最初定名為“dataXML”,其目的是要完全的取代前兩種數據標準。
制定mzML的最初設計方案是,讓mzML繼承mzData和mzXML各自的優點,并借鑒這兩種標準在實際使用時積累的經驗。在實現之初,開發人員遵從下面的設計原則:1)保持數據標準的簡潔;2)避免用不同的方法對同一信息進行編碼;3)為了可編碼一些新的重要信息,數據標準可以有一定的靈活性,但要保證標準的穩定性;4)繼承 mzData和mzXML的特色,但在最初版本中不需要太多體現;5)利用現有資源盡快完成初始版本(mzML 1.0);6)編寫讀、寫軟件驗證數據標準[28]。已發布的mzML主框架圖示于圖2。為了保證mzML能立即被廣泛應用,2008年6月發布mzML1.0時即向用戶提供了可以讀、寫和驗證的軟件。目前最新版本是2010年6月發布的mzML1.1.1。
2.1.4 3種數據標準的比較 mzData和mzXML最大的區別是數據文件靈活性的設計理念不同。mzData通過使用主體模型外部的控制字實現數據靈活性,這樣可以保證實際的xsd模型在幾年內保持不變。隨著設備和軟件的升級,只需不斷地更新控制字即可實現數據標準的升級,也無需過多修改后續的分析軟件。mzXML有一個嚴格的數據模型,大多數的數據信息都已被列舉在模型中,當需要支持新的數據特征時,即使添加一個屬性,均需要對數據模型進行修訂,也要對分析軟件進行相應的修改,造成連續發布的mzXML版本間多是相近的。但是,使用這種模型的優點是數據模型穩定性高,便于軟件的實現和數據文檔的驗證[20]。
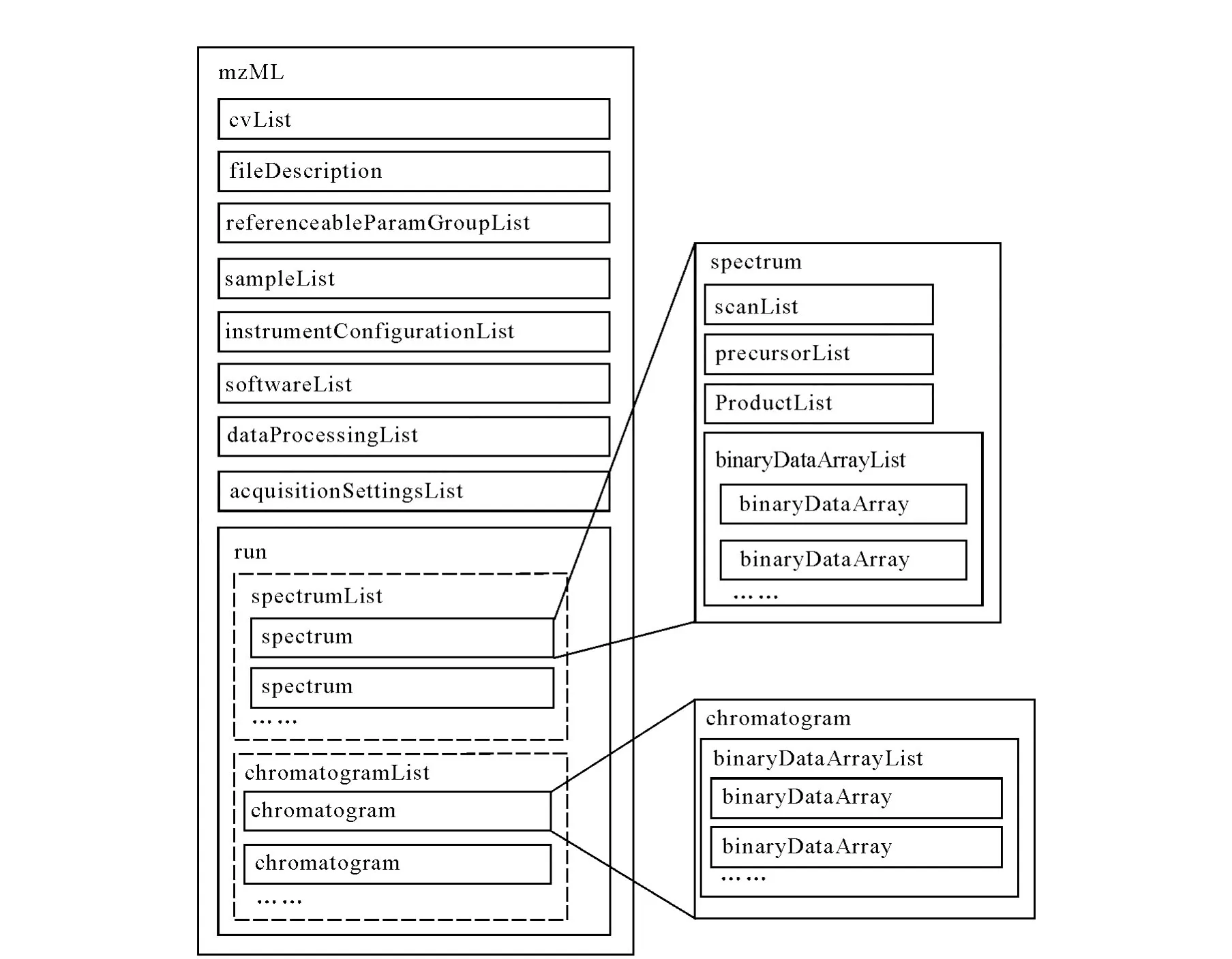
圖2 mzML結構主框圖Fig.2 The schema of mzML
mzML繼承了上述兩種格式各自的優點,使用控制字實現數據標準的靈活性,并使用控制字語法驗證器,避免用不同的方法編碼本質上相同的信息,以及各種版本間的不一致性,使mzML成為新一代基于 XML的開放式數據標準。
與mzXML相似,mzML使用索引來嘗試解決質譜圖隨機讀取的問題,但是該方法存在一定的風險。從mzXML多年的使用經驗可以得出,索引技術帶來的技術優勢要遠大于其帶來的風險,但研究人員對在XML文件中引入索引方法褒貶不一。因此,一個*.mzML文件中可能會包含一個無索引的mzML文檔或是帶索引的mzML文檔。
2.2 肽段水平分析階段
在蛋白質鑒定流程中,一個典型的肽段匹配策略是數據庫搜索[29],簡稱搜庫。目前 TPP中支持的搜庫工具有X!Tandem[30]、ProbID[31]、Mascot[32]、SEQUEST[33]、Phenyx[34]等 ,其中 ,X!Tandem和 ProbID是開源搜庫軟件。每個軟件都有各自的數據輸入輸出文件格式,而且在實際分析中,需要用不同的軟件對同一批數據進行搜庫,不同格式的輸出文件會對結果的比較、整合帶來一定困難,不利于大規模的數據分析。因此,研究人員在這一階段開發出的數據標準主要有:mzIdentML、pepXML和 Pride XML。其中,mzIdentML是針對該分析階段最新制定的數據標準,適用于搜庫后的數據分析流程。
2.2.1 mzIdentML的發展進程 2006年,PSIPI(proteomics informatics standards group)開始著手制定AnalysisXML的UML模型(unified modeling language model)[35]。隨后在2008年的 PSI春季會議上,決定從 AnalysisXML中去除與定量有關的部分,主要是因為定量中使用的不同策略(有標定量和無標定量)以及新技術的不斷更新,導致AnalysisXML1.0的發布一再推遲。2009年PSI的春季會議正式決定將AnalysisXML分成兩部分:蛋白質鑒定數據標準——mzIdentML和蛋白質定量數據標準——mzQuantML[16]。mzIdentML繼承了AnalysisXML的大部分內容,而mzQuantML還需要單獨開發,但仍期望在開發時使其具有與mzIdentML高度相似的上層結構。2009年 8月,mzIdentML1.0版正式發布,而 mzQuant-ML仍在制定中。
mzIdentML主要包含 pepXML和protXML中的數據信息,以及其他一些相關信息(與定量無關的)。pepXML是 TPP在肽段水平數據分析中使用的數據標準;protXML是TPP在蛋白質水平數據分析中使用的數據標準。因此,在基于質譜的蛋白質鑒定中,mzI-dentML是搜庫后結果的一個公共數據標準。TPP目前默認的數據標準還是pepXML,但在mzIdentML成熟后,TPP將會把最后的分析結果轉化成mzIdentML格式數據[36]。
2.2.2 mzIdentML的結構特點 mzIdentML是對 FuGE(functional genomics experiment)對象模型的延伸,可以應用于 MS、MS/MS、MSn數據的搜庫結果,比pepXML的適用性要廣。
mzIdentML同樣使用控制字來實現數據格式的靈活性,節點cvList中包含文件使用的控制字列表。節點Analysis Sample Collection使用控制字術語對試驗中質譜儀分析的樣本進行描述,若樣本是混合樣本,還需要定義其父樣本。Sequence Collection分為DBSequence和Peptide兩個子節點,前者為特定搜索數據庫(核酸或氨基酸)中的一個數據庫序列,后者為肽段序列(或修飾后序列),這兩個序列作為搜索結果的參考序列集。對數據集進行圖譜鑒定分析和蛋白質檢測所需要的參數和設置均存放在子節點Analysis Protocol Collection中,而分析過程中得到的數據結果存儲在 AnalysisData中。mzIdentML支持對數據的多次搜索,搜索結果可以存儲在同一個數據文件中。
mzIdentML數據標準適用于肽段水平分析和蛋白質水平分析,為整個蛋白質鑒定過程減輕了工作量,有效地避免了在分析中對不同數據格式進行轉換。
2.2.3 pepXML的結構特點 pepXML是ISB開發的用來存儲、交換和處理肽段序列匹配數據的數據標準,僅適用于MS/MS的搜庫結果。
在pepXML模型中,有相應的節點保證模型能支持搜庫、結果驗證和定量分析。其中,msms_pipeline_analysis存儲與搜庫有關的信息及搜庫結果;peptideprophet_summary和peptideprophet_result存儲與搜庫結果驗證有關的信息;asapratio_summary和asapratio_result存儲與ASAPRatio有關的定量信息。
pepXML支持在單文檔中存儲多次搜庫結果。每一次搜庫的結果都放在msms_run_summary中,其中包含原mzXML文件的信息和從mzXML文件得到質譜儀的詳細描述以及在試驗中使用的水解酶信息。此外還包含一個search_summary子節點,其中存儲與搜索引擎和搜索數據庫、肽段修飾、酶和序列搜索限制配置有關的信息,并將圖譜名、母離子電荷和質量以及節點search_result放在其子節點spectrum_query中。在對同一數據集做多次搜索時,每個search_result通過唯一的search_id與其對應的search_summary相連接,以實現在單文件中存儲多次搜索結果。
pepXML中還包含搜庫結果驗證和定量的模塊,如 TPP中使用的結果驗證軟件 PeptideProphet和定量軟件 XPRESS[37]、ASAPRatio[38],在pepXML中均有對應的數據模塊。
與SEQUEST的輸出文件SQL相比,pep-XML是一個有嚴格模型的XML文件格式,便于數據使用者驗證數據文件格式是否正確,保證分析軟件能有一個可靠的輸入數據。
2.2.4 Pride XML的結構特點 Pride XML是 EBI為數據庫PRIDE[39-41]專門開發的一個數據格式,其中包含了完整的圖譜數據以及搜庫結果。該標準在圖譜數據方面完全使用了mzData數據標準,將其作為一個節點 mzData。Pride XML中可以不包含mzData格式的圖譜數據,但在mzData節點中必須要對實驗樣品、儀器設備和數據處理軟件等參數進行詳細的描述。
Pride XML將基于不同實驗的搜庫結果分別存放在不同的節點中,二維凝膠電泳的搜庫結果存儲在 TwoDimensionalIdentification中,其它方法的搜庫結果存儲在 GelFreeIdentification中。同一肽段序列(或是重疊序列)的搜索結果存放在節點 GelFreeIdentification中,其中還需要包含的信息有搜索數據庫的名稱及版本、搜索引擎名稱及版本、相關圖譜ID、各項搜庫結果值以及修飾屬性等。
Pride XML同樣利用控制字來實現數據格式的靈活性,用戶可以在PRIDE的網站上對自己的XML文件進行驗證。
2.3 蛋白質水平分析階段
蛋白質鑒定過程的最后一步是通過搜庫中獲得的肽段結果推斷出樣品中含有的蛋白質。這一階段中已有的數據標準有mzIdentML和protXML,前者已在上一節做了介紹。
從得到的肽段序列推斷出蛋白質序列的方法有多種,不同的方法有不同格式的結果文件。protXML即是一個用于存儲、交換和處理基于串聯質譜的蛋白質鑒定結果的開放式數據標準。
protXML的節點protein_summary_header包含與肽段鑒定相關的信息,一個包含蛋白質鑒定方法信息的子節點program_details和一個包含該方法細節信息的通配符。蛋白質鑒定結果存放在protein_group中,可以有多組,每組中均有一個編號group_number,每組中還可以包含一個或多個protein節點。protXML也適用于鑒定結果的后續蛋白質水平上的分析,如XPRESS和 ASAPRatio蛋白質定量。protXML文件還分別設有對應的節點存儲相應的分析結果,如ASAPRatio的定量結果存儲在節點ASAPRatio中。
3 支持數據標準的軟件
上述數據標準自發布之日起便逐步得到了應用,常用的蛋白質組質譜分析軟件通常都支持多種數據標準,表3給出了常用軟件對數據標準的支持情況。
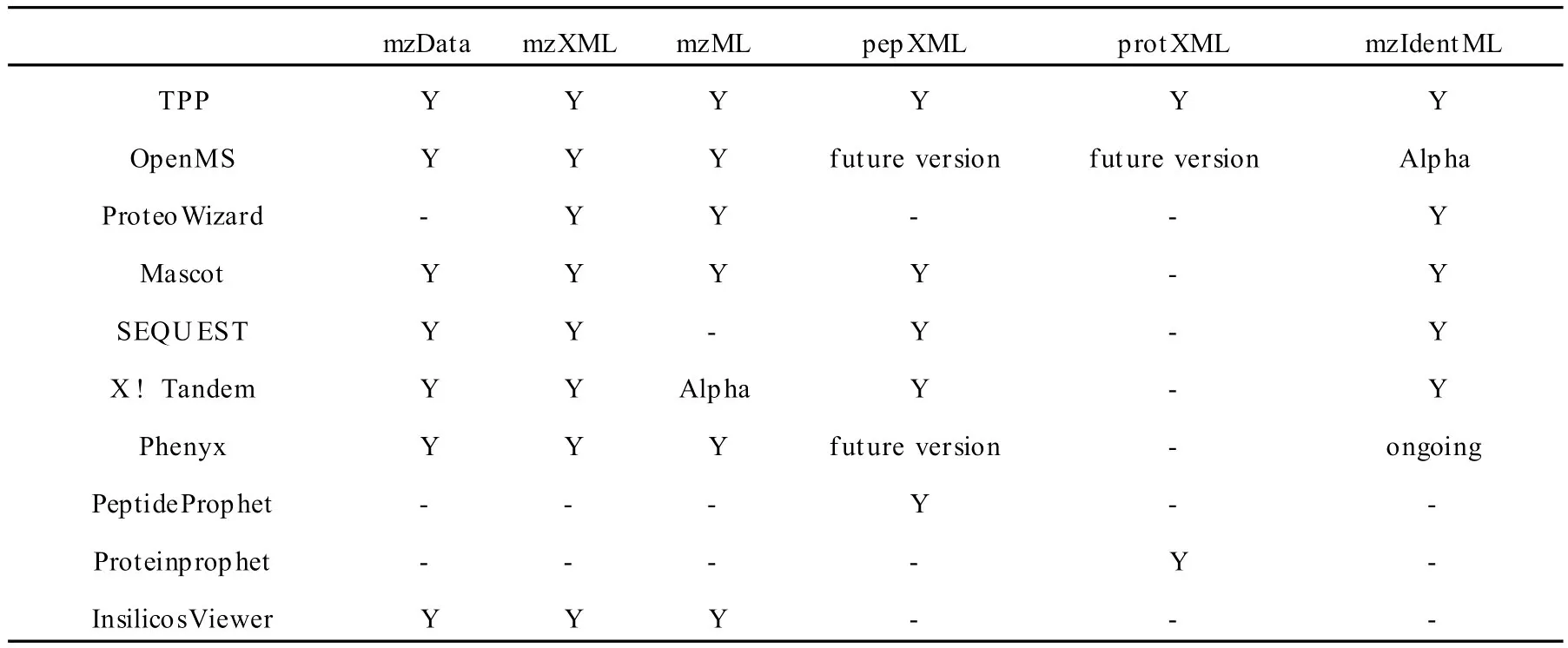
表3 常用支持數據標準的軟件列表Table 3 Widely-used softwares and their support for the XML-based data format standards
4 基于XML數據標準的優缺點
基于XML的質譜數據標準在質譜數據分析中發揮了極大的作用,減輕了分析人員在數據分析中對不同格式的數據進行轉換的工作,有利于大規模數據分析平臺的建設。但這些數據標準還存在一定的問題,需要不斷的完善和發展。
首先,已有標準對原始數據共享的支持力度比較大,但是對數據分析結果的支持有限。ISB制定的pepXML和protXML,以及 EBI的Pride XML雖然提供了部分對搜庫結果存儲的支持,但是還不能兼容所有出現的、典型的數據分析流程。
其次,這些數據標準多以靈活的XML格式實現,其中大量的數據標簽使容量本已足夠龐大的高通量質譜數據文件更加龐大,數據壓縮雖然可以部分解決這個問題[42-43],但是不能兼顧數據的訪問速度。
最后,XML平坦式的存儲方式給數據的高效訪問帶來了一定負擔,通過建立額外的索引雖可以部分解決這一問題,但是直接的索引式存儲效率應更加高效。
5 展望
質譜數據具有數據量大和數據格式不統一的特點,研究工作的需求,牽引和推動了數據標準的發展,但目前數據標準的制定仍然落后于數據的發展。通過比較目前已有的數據標準并結合實際數據處理的諸多經驗,建議從兩個不同的層面關注和推動今后數據標準的發展:
1)對數據標準的制定組織,仍需繼續完善和發展蛋白質組學中所需數據標準。上文介紹的很多數據標準僅僅適用于蛋白質鑒定的典型流程,使用范圍有限。因此,數據標準的制定還需要不斷完善。從各數據標準的發展過程以及mzML與mzIdentML的制定過程可以推斷出質譜數據標準在向一個可以支持所有蛋白質組學典型分析流程的方向發展。由于蛋白質組學自身的特點,如實驗策略多、分析步驟復雜以及數據分析算法繁多等,使得這一發展過程困難重重。
2)對數據分析人員,在目前數據標準不統一的狀況下,可開發基于已有數據標準的適用于典型蛋白質鑒定流程的數據格式,并充分利用已有的開源數據格式轉換工具,將當前各種不同數據標準的數據信息合理地整合到一個統一的數據格式中,以簡化數據分析流程。在此實踐過程中,可以向數據標準制定組織及時反饋使用經驗,共同推動蛋白質組學中數據標準的發展。
[1]Science Editor.Breakthrough of the year.Peering into 2002[J].Science,2001,294(5 551):2 444.
[2]AEBERSOLD R,MANN M.Mass spectrometrybased proteomics[J].Nature,2003,422(6 928):198-207.
[3]ONG S E,MANN M.Mass spectrometry-based proteomics turns quantitative[J].Nat Chem Biol,2005,1(5):252-262.
[4]STATES D J,OMENN G S,BLACKWELL T W,et al.Challenges in deriving high-confidence protein identifications from data gathered by a HUPO plasma proteome collaborative study[J].Nat Biotechnol,2006,24(3):333-338.
[5]The HUPO Proteomics Standards Initiative[OL][2010].http://www.psidev.info/.
[6]Institute for System Biology[OL][2010].http://www.systemsbiology.org/.
[7]European Bioinformatics Institute[OL][2010].http://www.ebi.ac.uk/.
[8]KA ISER J.Proteomics:Public-private group maps outinitiatives[J]. Science,2002,296(5 569):827.
[9]ACHARD F,VAYSSEIX G,BARILLOT E.XML,bioinformatics and data integration[J].Bioinformatics,2001,17(2):115-125.
[10]BRAY T,PAOLI J,SPERBERG-MCQUEEN C M,et al.Extensible Markup Language(XML)1.0(Second Edition),2000.
[11]TAYLOR C F,PATON N W,LILLEY KS,et al.The minimum information about a proteomics experiment(MIAPE)[J].Nat Biotechnol,2007,25(8):887-893.
[12]TAYLOR C F,BINZ P A,AEBERSOLD R,et al.Guidelines for reporting the use of mass spectrometry in proteomics[J]. NatBiotechnol,2008,26(8):860-861.
[13]ORCHARD S,TAYLOR C F,HERMJAKOB H,et al.Advances in the development of common interchange standards for proteomic data[J].Proteomics,2004,4(8):2 363-2 365.
[14]PEDRIOLI P G,ENGJ K,HUBLEY R,et al.A common open representation of mass spectrometry data and its application to proteomics research[J].Nat Biotechnol,2004,22(11):1 459-1 466.
[15]VIZCAINO J A,MARTENS L,HERMJAKOB H,et al.The PSI formal document process and its implementation on the PSI website[J].Proteomics,2007,7(14):2 355-2 357.
[16]ORCHARD S,DEUTSCH E W,BINZ P A,et al.Annual spring meeting of the Proteomics Standards Initiative[J].Proteomics,2009,9(19):4 429-4 432.
[17]LIN S M,ZHU L,WINTER A Q,et al.What is mzXML good for?[J].ExpertRev Proteomics,2005,2(6):839-845.
[18]PSI-MS:Mass Spectrometry Standards Working Group[OL][2010].http://www.psidev.info/index.php?q=node/80.
[19]ORCHARD S,MONTECHI-PALAZZIL,DEUTSCH E W,et al.Five years of progress in the standardization of proteomics data 4th annual spring workshop of the HUPO-proteomics standards initiative April 23-25,2007 ecolenationalesuperieure(ENS),Lyon,France[J].Proteomics,2007,7(19):3 436-3 440.
[20]DEU TSCH E.mzML:A single,unifying data format for mass spectrometer output[J].Proteomics,2008,8(14):2 776-2 777.
[21]PRoteomics IDEntifications database(PRIDE)[J/OL][2010].http://www.ebi.ac.uk/pride/.
[22]Seattle proteome center(SPC)-Proteomics Tools[EB/OL].http://tools.proteomecenter.org/software.php.
[23]PRINCE J T,CARL SON M W,WAN G R,et al.The need for a public proteomics repository[J].Nat Biotechnol,2004,22(4):471-472.
[24]CARR S,A EBERSOLD R,BALDWIN M,et al.The need for guidelines in publication of peptide and protein identification data:Working group on publication guidelines for peptide and protein identification data[J].Mol Cell Proteomics,2004,3(6):531-533.
[25]ORCHARD S,HERMJAKOB H,JULIAN R K,et al.Common interchange standards for proteomics data:Public availability oftools and schema[J].Proteomics,2004,4(2):490-491.
[26]KELL ER A,ENGJ,ZHANG N,et al.A uniform proteomics MS/MS analysis platform utilizing open XML file formats[J].Mol Syst Biol,2005:0017.
[27]ORCHARD S,HERMJAKOB H.The HUPO proteomics standards initiative-easing communication and minimizing data loss in a changing world[J].Brief Bioinform,2008,9(2):166-173.
[28]DEUTSCH E W.Mass spectrometer output file format mzML[J].Methods Mol Biol,2010,604:319-331.
[29]XU C,MA B.Software for computational peptide identification from MS-MS data[J].Drug Discov Today,2006,11(13/14):595-600.
[30]CRAIG R,BEAVIS R C.TANDEM:Matching proteins with tandem mass spectra[J].Bioinformatics,2004,20(9):1 466-1 467.
[31]ZHANG N,AEBERSOLD R,SCHWIKOWSKI B.ProbID:A probabilistic algorithm to identify peptides through sequence database searching using tandem mass spectral data[J].Proteomics,2002,2(10):1 406-1 412.
[32]PERKINS D N,PAPPIN D J,CREASY D M,et al.Probability-based protein identification by searching sequence databases using mass spectrometry data[J]. Electrophoresis,1999,20(18):3 551-3 567.
[33]ENGJ K,MCCORMACK A L,IIIJRY.An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database[J].Journal of the American Society for Mass Spectrometry,1994,5(11):976-989.
[34]COLINGE J,MASSELOT A,GIRON M,et al.OLAV:Towards high-throughput tandem mass spectrometry data identification[J].Proteomics,2003,3(8):1 454-1 463.
[35]ORCHARD S,APWEILER R,BARKOVICH R,et al.Proteomics and Beyond:A report on the 3rd annual spring workshop of the HUPO-PSI 21-23 April 2006,San Francisco,CA,USA[J].Proteomics,2006,6(16):4 439-4 443.
[36]DEUTSCH E W,MENDOZA L,SHTEYNBERG D,et al.A guided tour of the trans-proteomic pipeline[J].Proteomics,2010,10(6):1 150-1 159.
[37]HAN D K,ENGJ,ZHOU H,et al.Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry[J].Nat Biotechnol,2001,19(10):946-951.
[38]LI XJ,ZHANG H,RANISH J A,et al.Automated statistical analysis of protein abundance ratios from data generated by stable-isotope dilution and tandem mass spectrometry[J].Anal Chem,2003,75(23):6 648-6 657.
[39]MARTENS L,HERMJA KOB H,JONES P,et al.PRIDE:The proteomics identifications database[J].Proteomics,2005,5(13):3 537-3 545.
[40]JONES P,COTE R G,MARTENS L,et al.PRIDE:A public repository of protein and peptide identifications for the proteomics community[J].Nucleic Acids Res,2006,34(suppl 1):D659-D663.
[41]JONES P,COTE RG,CHO S Y,et al.PRIDE:New developments and new datasets[J].Nucleic Acids Res,2008,36(suppl 1):D878-D883.
[42]MIGUEL A C,KEANE J F,WHITEAKER J,et al.Compression of LC/MS Proteomic data[C].19th IEEE Symposium on Computer-Based Medical Systems, Salt Lake City, 2006:925-930.
[43]MIGUEL A C,KEARNEY-FISCHER M,KEANE J F,et al.Near-lossless compression of mass spectra for proteomics[C].Acoustics,Speech,and Signal Processing,Honolulu,2007:1 369-1 372.
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
當代陜西(2019年8期)2019-05-09 02:22:48
上海建材(2019年1期)2019-04-25 06:30:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
質量與標準化(2015年9期)2015-12-31 11:41:40
中國質量與標準導報(2014年4期)2014-03-11 19:54:25
中國質量與標準導報(2014年10期)2014-02-28 22:25:47
中國質量與標準導報(2014年7期)2014-02-28 22:24:39
