全外顯子組測序技術及其在腫瘤研究中的應用
2011-01-24 02:13:30陳怡然吳小華
中國腫瘤外科雜志 2011年3期
關鍵詞:研究
陳怡然, 吳小華
隨著2002年人類基因組計劃的完成,以及其他種類生物全基因組圖譜的陸續(xù)描繪,發(fā)現不同種群間的基因差別僅為1%,主要表現為外顯子的差異。外顯子作為蛋白質的編碼區(qū),是DNA中的重要功能序列,其僅占人類基因組約1%[1],但涵蓋了與個體表型相關的大部分功能性變異,與人類約85%的導致疾病的基因突變相關[2]。全外顯子組(exome)為人類基因組全部外顯子區(qū)域的總稱,全外顯子組測序(whole-exome sequencing)又稱為定向外顯子組捕獲(targeted exome capture),可選擇性的對人類基因組的編碼區(qū)進行測序,從而發(fā)現罕見及常見疾病相關的異常基因[3]。目前,全外顯子組測序相比于全基因組測序,其低成本、高效率的優(yōu)勢日益顯著。在單基因疾病的應用中,其不僅加快單基因缺陷疾病的基因確定,在系統(tǒng)地預測疾病相關基因方面亦有應用。對于復雜疾病如腫瘤、糖尿病等的致病基因和易感基因的測定等[4]也有相當的應用前景。本文主要對全外顯子組測序技術以及目前該技術在腫瘤研究中的應用等問題進行闡述。
1 全外顯子組測序技術
全外顯子組測序技術作為一種新型的基因組分析技術,包括全外顯子組序列捕獲及測序。
1.1 全外顯子組序列捕獲
以兩種較成熟的方法為例。
1.1.1NimbleGen外顯子序列捕獲芯片法基因組DNA被隨機打斷成片段,隨后在DNA片段兩端分別連上接頭,與定制的序列捕獲芯片雜交,去除未與芯片結合的背景DNA,將經過富集的外顯子區(qū)域的DNA洗脫并擴增。
1.1.2In-Solution 捕獲法所要求的基因起始量低,能有效地捕獲包含未知突變的DNA。具體步驟[5]:(1)基因組DNA被打斷,并組裝成測序平臺特異的文庫形式。在捕獲之前,對文庫的大小進行選擇,并利用電泳等方法來進行驗證。(2)精選大小的文庫,隨后與誘餌共同孵育24 h。(3)加入鏈酶親和素標記的磁珠,并通過強磁鐵從混合物中釣出RNA誘餌-DNA雜合體。(4)洗脫磁珠,將RNA誘餌降解,將得到的目的DNA進行常規(guī)的擴增和測序。
以上方法主要通過捕獲芯片或溶液型目標富集系統(tǒng)完成。捕獲芯片作為定向測序的快速有效的方法,更適用于小型化研究,而溶液型則適用于樣品數量上千的大規(guī)模高通量測序研究。
1.2 全外顯子組測序
目前應用的主要測序平臺有:Roche454,Illumina Genome Analyzer Ⅱ,Applied Biosystems SOLiD。
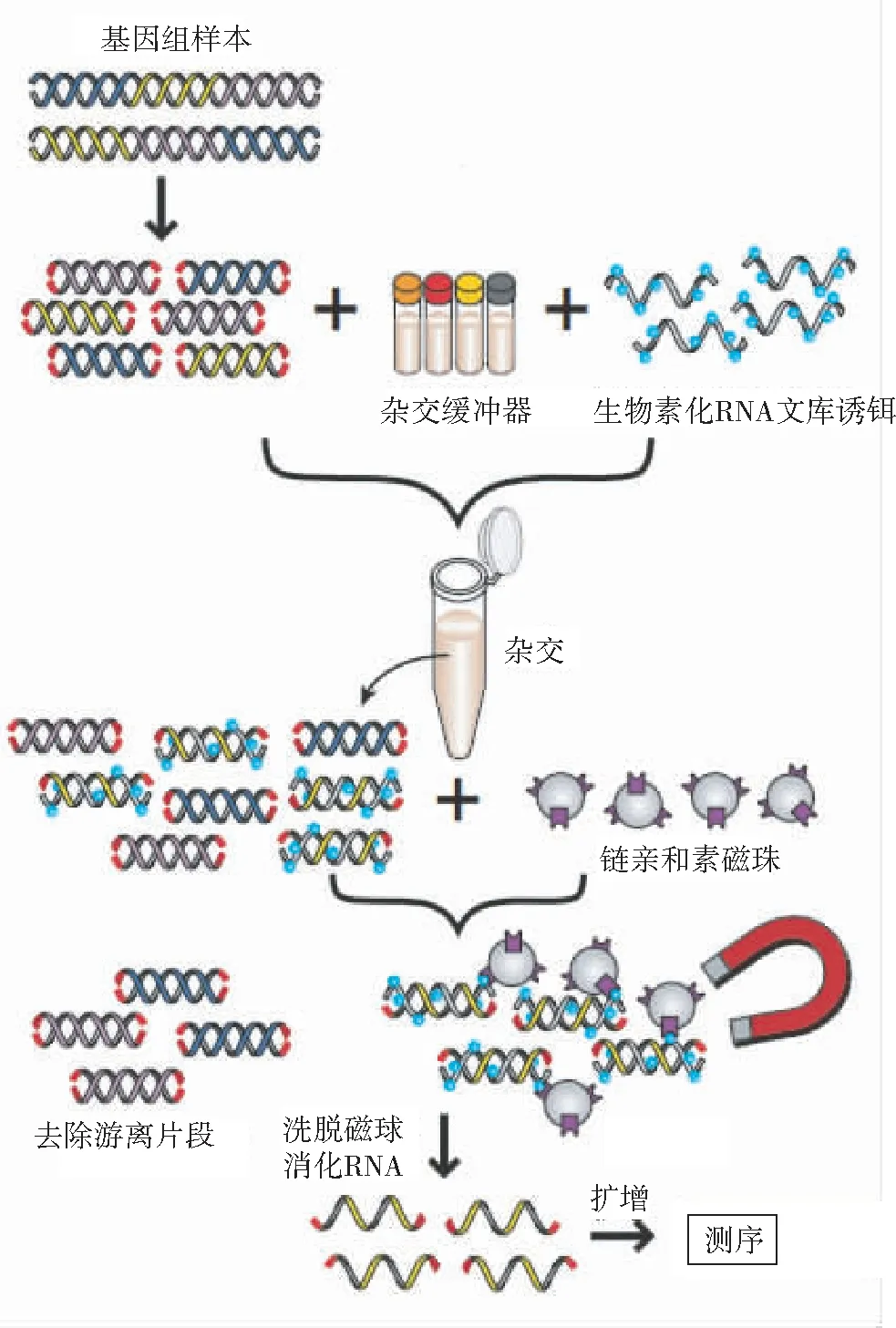
圖1 In-Solution法捕獲外顯子組流程[5]
1.2.1Roche454即大規(guī)模并行焦磷酸合成測序法,采用GS FLX系統(tǒng):每個磁珠通過接頭,與一條大小為300~800bp的DNA片段結合構成樣品文庫。包含磁珠和擴增試劑的水溶液注入礦物油中,形成油包水的乳濁液結構,乳濁液PCR在每個小水滴中獨立進行,產生幾百萬相同的拷貝,焦磷酸測序反應測得每個磁珠上的片段產生一條讀長,由此進行數據分析[6]。
1.2.2Applied Biosystems SOLiD即基于磁珠的大規(guī)模并行克隆連接DNA測序法,采用SOLD系統(tǒng):利用微珠通過接頭捕獲DNA片段,之后進行乳液PCR。其以連接反應取代傳統(tǒng)的聚合酶延伸反應,以8bp的探針作為連接反應的底物,探針設計為將3′端1~2位的堿基與5′端標記的熒光信號的顏色相對應。一次SOLiD測序共為五輪循環(huán)測序,每輪由多個連接反應組成。五輪測序完成后,可得到各位置的顏色信息,從而推斷出相應的堿基序列[7]。
1.2.3Illum ina Genome Analyzer Ⅱ即合成測序法,采用GA Ⅱ系統(tǒng)。單鏈的DNA片段兩端通過接通固定到芯片表面,形成橋結構,之后以寡核苷酸為引物進行PCR擴增,得到單克隆DNA族群。測序中,利用“可逆終止子”,使得每個測序循環(huán)只摻入單個堿基,獲得各位置單個堿基的信息后,將反應試劑洗脫,加入新的反應體系,開始下一測序循環(huán)[8]。
新一代的測序平臺,與傳統(tǒng)測序技術相比,利用接頭構建基因文庫,在反應基質上進行大規(guī)模的并行PCR,實現了邊合成邊測序的可能性。
不可否認的是,全外顯子測序技術作為一種新型技術,目前其對于全基因組蛋白質編碼區(qū)的覆蓋并不如設計的理想,Jacobs等[9]評估后發(fā)現,NimbleGen的捕獲探針捕獲蛋白質編碼序列堿基率為77%,Agilent為83%,并且不同方法的測序亦均有一定程度的缺失。故目前對于該新型技術的應用,尤其是對于實驗陰性結果的解釋,仍需謹慎為之。
2 全外顯子組測序技術在腫瘤學方面的應用現狀
2.1 找尋潛在癌基因及抑癌基因,為腫瘤的分子診斷提供參考
全外顯子組測序技術由于對外顯子的高覆蓋而獲得大量的基因樣本。故即使在樣本中腫瘤細胞含量較少和(或)樣本純度差異較大的情況下,其仍可較好的對外顯子區(qū)域的基因突變進行測定,從而進一步確定腫瘤相關癌基因及抑癌基因,為腫瘤的早期診斷提供依據[10]。
Wei等[11]對14位配對的正常人及轉移黑素瘤患者進行比較研究,發(fā)現TRRAP頻發(fā)突變具有致癌功能;患者GRIN2A突變頻率達33%;突變均發(fā)生在谷氨酸信號通路的編碼區(qū)域,通過該信號通路的修飾達到抑制腫瘤生長成為潛在的治療手段。類似地,Varela等[12]在對7例腎透明細胞癌患者的研究中,發(fā)現SWI/SNF染色質重組復合體基因PBRM1作為該腫瘤的第二大癌基因,在患者組截斷突變率達41%。Jones等[13]通過對42例卵巢透明細胞癌患者的腫瘤和正常細胞的對比分析,篩選得到4個基因,PIK3CA編碼磷脂酰肌醇-3激酶亞單位,KRAS與已知癌基因產物相關。PPP2R1A編碼絲氨酸-蘇氨酸磷酸酶2的調控亞單位,功能類似癌基因。ARID1A作為腫瘤抑制基因編碼產物參與染色體重構,并且其在42例患者中突變率達到57%。Parsons等[14]在兒童髓母細胞瘤的研究中,發(fā)現16%的髓母細胞瘤的患者中存在著組蛋白-賴氨酸-N甲基轉移酶基因MLL2或MLL3的鈍化突變。該研究同時揭示了兒童及成人髓母細胞瘤的遺傳景觀上的關鍵差異。
相比于散發(fā)而言,家族性腫瘤的遺傳傾向常更明顯,故全面的、無偏倚的蛋白質編碼區(qū)的基因測序成為確定相關責任基因的一種可行方法。Jones等[15]對家族性胰腺癌進行測定,發(fā)現該家系的3個獨立的截斷突變,確立了PALB2為家族性胰腺癌的易感基因。Goldin等[16]在對腫瘤易感家族的研究中,對3個黑色素瘤及2個霍杰金淋巴瘤的家族以及各家族1名遠親對比進行全外顯子組的測序,發(fā)現每個家族可以獲得0~100不等的候選非同義突變。通過追蹤,對各家族其他成員進行基因分析,使得篩選該腫瘤的家族易感基因成為可能。
2.2 協(xié)助腫瘤分型
某些特定腫瘤的基因分型,對之后的治療方案、靶向藥物的選擇和治療結局等都有一定的指示作用,故利用適當的基因測序技術,對其進行個體化分型,成為未來腫瘤治療的一大趨勢。Timmermann等[17]發(fā)現,由于全外顯子組測序可以平行探測微衛(wèi)星不穩(wěn)定性(microsatellite instability,MSI) 結直腸癌的頻發(fā)突變,潛在錯配修復(mismatch repair, MMR)機制的缺失甚至對結直腸癌亞型的高頻突變如BRAF、KRAS和TP53進行測定,使其可能成為未來結直腸癌基因分型的工具。
2.3 腫瘤轉移遺傳學
轉移作為惡性腫瘤的一大特征,是腫瘤患者死亡的最常見原因,但目前對于腫瘤轉移遺傳學的所知甚少。Harbour等[18]經過對轉移性的葡萄膜黑素瘤進行外顯子組捕獲及高通量平行測序,發(fā)現BAP1(BRCA1-關聯蛋白1)的缺失與該病高轉移性相關。另外,胰腺癌預后不佳常常與其不能早期診斷及早期遠處轉移相關。Yachida等[19]報道,對7例轉移胰腺癌患者進行基因測序,除評估原發(fā)灶與轉移灶之間同源細胞關系之外,亦對胰腺癌的遺傳學演變進行 了研究,發(fā)現細胞原始突變的發(fā)生至原位腫瘤親代細胞的產生至少需要10年時間,之后獲得轉移能力至少經歷5年,轉移后患者平均在2年內死亡。其結論提示,可以通過研究腫瘤進展的遺傳學特征,明確腫瘤轉移前的時間窗,以期早期發(fā)現,預防腫瘤轉移導致的死亡結局[20]。
2.4 選擇腫瘤治療的相關靶點
Pasqualucci等[21]關于B細胞非霍奇金淋巴瘤中最常見類型——彌漫大B細胞淋巴瘤以及濾泡性淋巴瘤的研究中發(fā)現,基因組結構的改變使得與組蛋白及非組蛋白乙酰轉移酶高度相關的基因CREBBP和EP300鈍化,其表達不足造成BCL6和p53的乙酰化不足,最終導致相關腫瘤蛋白的激活以及p53腫瘤抑制基因活性的下調,提示可利用藥物針對乙酰化/去乙酰化進行靶向治療。Timmermann等[17]研究顯示,結直腸癌中BRAF、KRAS、FGFR2、MTOR等腫瘤藥物靶區(qū)的基因突變,直接影響腫瘤藥物的治療效果。Parsons等[14]在兒童髓母細胞瘤的研究中,發(fā)現一種特別類型的組蛋白甲基化在該腫瘤形成中的作用,提供靶向抑瘤的研究方向。Jiao等[22]在胰腺內分泌腫瘤的研究中,除了發(fā)現關于DAXX、ATRX、MEN1等突變導致的染色體重組,亦顯示14%的該腫瘤患者存在鈉巴霉素的靶向通路(mammalian target of rapamycin, mTOR)相關基因的突變,從而使得篩選該類患者進行mTOR抑制治療存在可能。
因此,通過全外顯子組測序技術獲得腫瘤基因突變相關的最佳藥物靶點,改變相應代謝途徑,或可能成為未來腫瘤個體化治療的金標準。
2.5 提示腫瘤預后
Yan等[23]研究顯示, 在急性單核細胞性白血病(AML-M5)患者中,DNMT3A 基因突變導致了DNA甲基轉移酶的活性降低,并表現出對組蛋白H3的異常親和力。DNA甲基化作為重要的基因組修飾,調控著許多重要細胞過程,其與腫瘤發(fā)生的相關性也已被證實。研究者經分析后發(fā)現DNMT3A作為與總生存率相關的獨立預后變量,攜帶該基因常顯示預后不佳。而Jiao等[22]關于胰腺內分泌腫瘤臨床研究中顯示,DAXX、ATRX、MEN1基因突變則提示較好預后。
2.6 應用于干細胞研究
腫瘤干細胞(Cancer Sterm Cells, CSCs)學說認為腫瘤中存在著一類細胞,其抗調亡能力、成瘤能力、遷徙和侵襲能力等與占大多數分化程度較高的腫瘤細胞存在差異[24],是腫瘤無限增殖、復發(fā)和轉移的根源[25]。該理論為腫瘤的發(fā)生、發(fā)展、復發(fā)和轉移等機制的研究提供了全新的視角。目前,全外顯子組測序技術應用于干細胞,甚至是腫瘤干細胞的研究尚處于初步階段。Howden等[26]在對于1例視網膜環(huán)狀萎縮患者誘導性多潛能干細胞(IPS cell)的遺傳校正及分析研究中,對IPS細胞株進行最初的重編程、基因靶向、選擇性片段移除后,聯合應用全外顯子組測序技術與aCGH方法,評估其基因組完整性。找到該患者原始IPS細胞的蛋白質編碼區(qū)域中共有1個基因缺失、2個基因擴增以及9個基因突變。為研究該疾病IPS細胞分化潛能的機制提供了新思路。Totoki等[27]在關于肝細胞癌的基因組研究顯示,發(fā)現的47個非同義突變中的TSC1復合體的無義突變僅在小部分的腫瘤細胞中出現,并與負性調節(jié)鈉巴霉素的哺乳動物靶向通路有關,成為與該腫瘤細胞的“干性”表達、生長、代謝相關的重要致癌途徑。
綜上所述,目前全外顯子組測序技術在腫瘤學的應用仍處于較早期的階段。相比于傳統(tǒng)全基因組測序技術,其低花費、高產量已成為明顯的優(yōu)勢,在腫瘤分子生物學的研究和臨床應用方面顯示了多方面的影響。相信隨著測序成本的進一步降低,通過該技術對腫瘤進行基因水平的診斷、分型,治療靶點的確立以及預后的分析等,將成為未來腫瘤分子生物學及個體化治療的有效手段。
參考文獻:
[1]Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes[J]. Nature, 2009,461(7261): 272-276.
[2]Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing[J]. Proc Natl Acad Sci U S A, 2010, 106(45): 19096-19101.
[3]Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a mendelian disorder[J]. Nat Genet, 2010, 42(1): 30-35.
[4]Lehne B, Lewis CM, Schlitt T.Exome localization of complex desease association signals[J]. BMC Genomics, 2011, 12:92.
[5]Ernani FP, LeProust EM. Agilent’s sureselect target enrichment systems: Bringing cost and process efficiency to next-generation sequencing[EB/OL]. http://www.chem.agilent.com/Library/brochures/5990-3532en_lo%20CMS.pdf
[6]Margulies M, Egholm M, Altman WE, et al.Genome sequencing in open microfabricated high density picolitre reactors[J]. Nature, 2005, 437(7057): 376-380.
[7]Smith DR, Quinlan AR, Peckham HE, et al. Rapid whole-genome mutational profiling using next-generation sequencing technologies[J]. Genome Res, 2008,18(10): 1638-1642.
[8]Bentley DR, Balasubramanian S, Swerdlow HP,et al. Accurate whole human genome sequencing using reversible terminator chemistry[J]. Nature, 2008, 456(7218): 53-59.
[9]Jacobs KB, Yeager M, Cullen MG, et al. Realities and limitations of coverage in current whole-exome sequencing capture approaches[J]. Genetic epidemiology, 2010, 34(8): 919-920.
[10]Meyerson M, Gabriel S, Getz G.Advances in understanding cancer genomes through second-generation sequencing[J]. Nat Rev Genet, 2010, 11(10): 685-696.
[11]Wei X, Walia V, Lin JC,et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma[J]. Nat Genet, 2011,43(5):442-446.
[12]Varela I, Tarpey P, Raine K,et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma[J]. Nature, 2011, 469(7331): 539-542.
[13]Jones S, Wang TL, Shih IeM,et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma[J]. Science, 2010,330(6001):228-231.
[14]Parsons DW, Li M, Zhang X,et al. The genetic landscape of the childhood cancer medulloblastoma[J]. Science, 2011, 331(6016):435-439.
[15]Jones S, Hruban RH, Kamiyama M,et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene[J]. Science, 2009, 324(5924): 217.
[16]Goldin LR, Yang XR, McMaster ML, et al. The use of whole exome sequencing to identify rare susceptibility variants in cancer prone families[J]. Genetic epidemiology,2010, 34(8):924.
[17]Timmermann B, Kerick M, Roehr C, et al. Somatic mutation profiles of MSI and MSS colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis[J]. PLoS One, 2010, 5(12): e15661.
[18]Harbour JW, Onken MD, Roberson ED,et al. Frequent mutation of BAP1 in metastasizing uveal melanomas[J]. Science, 2010,330(6009): 1410-1413.
[19]Yachida S, Jones S, Bozic I,et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer[J]. Nature, 2010,467(7319):1114-1117.
[20]Campbell PJ, Yachida S, Mudie LJ, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer[J]. Nature, 2010, 467(7319): 1109-1113.
[21]Pasqualucci L, Dominguez-Sola D, Chiarenza A, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma[J]. Nature, 2011,471(7337): 189-195.
[22]Jiao Y, Shi C, Edil BH, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors[J]. Science, 2011, 331(6021):1199-1203.
[23]Yan XJ, Xu J, Gu ZH,et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia[J].Nature Genetics, 2011,43(4): 309-315.
[24]O’Brien CA, Kreso A, Jamieson CH. Cancer stem cells and self-renewal[J]. Clin Cancer Res. 2010, 16(12): 3113-3120.
[25]Marx J. Cancer research. Mutant stem cells may seed cancer[J]. Science, 2003, 301(5638): 1308-1310.
[26]Howden SE, Gore A, Li Z, et al. Genetic correction and analysis of induced pluripotent stem cells from a patient with gyrate atrophy[J]. Proc Natl Acad Sci U S A,2011, 108(16): 6537-6542.
[27]Totoki Y, Tatsuno K, Yamamoto S et al. High-resolution characterization of a hepatocellular carcinoma genome[J]. Nat Genet, 2011, 43(5): 464-469.
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
