乙醇胺為堿性催化劑對硅氣凝膠性能的影響*
2011-01-22 02:17:49,,
無機(jī)鹽工業(yè) 2011年9期
關(guān)鍵詞:催化劑
,,
(1.新疆大學(xué)化學(xué)化工學(xué)院,新疆烏魯木齊 830046;2.新疆烏魯木齊市氣象局)
二氧化硅氣凝膠具有高比表面積、高孔隙率、低密度、低熱導(dǎo)率、極低介電系數(shù)和低折射系數(shù)的特殊性能,同時綠色環(huán)保,在熱學(xué)、光學(xué)、化工等方面具有廣泛的商業(yè)應(yīng)用價值。制備硅氣凝膠硅源一般采用正硅酸四乙酯(TEOS),酸催化劑一般采用HF、鹽酸、乙酸、草酸[1]等,堿性催化劑通常采用氨水。干燥方法為超臨界干燥法和常壓干燥法。常壓干燥法是今后的發(fā)展方向,但表面改性劑的使用條件較為苛刻[2]。S.Yoda等以乙醇胺為催化劑,直接加入到正硅酸四甲酯(TMOS)中,使TMOS凝膠,結(jié)果表明乙醇胺的加入對膠凝時間、收縮率、孔隙率等均有影響[3]。F.Carre等認(rèn)為醇胺直接與硅烷中的硅發(fā)生配位反應(yīng),硅醇的—OR基團(tuán)阻止了水解反應(yīng)[4]。直到目前,硅氣凝膠仍不具有商業(yè)價值,主要是受到硅源和表面改性劑高昂價格的限制[5]。乙醇胺同時具有胺基和羥基結(jié)構(gòu),其對氣凝膠的合成有影響。筆者以鹽酸為酸催化劑,分別以一乙醇胺(MEA)、二乙醇胺(DEA)、三乙醇胺(TEA)為堿性催化劑,并與氨水作比較,采用溶膠-凝膠法合成濕凝膠,不經(jīng)過表面改性,采用真空干燥得到二氧化硅氣凝膠。通過對所得氣凝膠的結(jié)構(gòu)特征進(jìn)行表征,表明乙醇胺為堿性催化劑具有不同于氨水的性質(zhì)。
1 實驗
1.1 SiO2氣凝膠的制備
以TEOS、蒸餾水為原料,無水乙醇(EtOH)為溶劑,鹽酸為酸催化劑,乙醇胺(MEA、DEA、TEA)、氨水為堿催化劑,采用酸堿兩步催化溶膠-凝膠共前驅(qū)體法制備二氧化硅濕凝膠。通過對主要合成參數(shù)的優(yōu)化,得到TEOS、EtOH、水物質(zhì)的量比為1∶4∶4。加入鹽酸(5.3×10-3mol)、堿性催化劑[MEA、DEA、TEA、氨水,(7.6~85)×10-3mol]進(jìn)行凝膠聚合反應(yīng)。將凝膠置于60 ℃水浴中老化,期間加入無水乙醇置換原溶劑,每隔24 h替換一次無水乙醇,老化總時間為3~4 d。將濕凝膠在真空條件下采用分段升溫方式每隔5 ℃從40 ℃升溫至70 ℃進(jìn)行干燥,得到干燥SiO2氣凝膠。
1.2 SiO2氣凝膠的表征
使用精度為0.000 1 g的電子天平稱量已知體積的氣凝膠質(zhì)量,根據(jù)下式估算孔隙率:
孔隙率=(1-ρp/ρs)×100%
(1)
式中:ρp為氣凝膠表觀密度,g/cm3;ρs為SiO2理論密度,2.2 g/cm3。
采用德國BRUKER公司的EQUINOX型傅里葉變換紅外光譜儀對樣品進(jìn)行紅外光譜測試;采用德國LEO公司的LEO1430VP型環(huán)境掃描電子顯微鏡觀察SiO2氣凝膠的微觀形貌和孔隙結(jié)構(gòu);采用中國產(chǎn)JW-BK型靜態(tài)氮吸附儀,采集脫吸附曲線數(shù)據(jù)并利用BET原理計算氣凝膠的比表面積;采用BJH計算方法求得氣凝膠的孔徑分布和平均孔徑,測試前樣品先在105 ℃熱處理2 h,測試溫度為-195.8 ℃。
2 結(jié)果與討論
2.1 SiO2氣凝膠吸附性能比較
根據(jù)BET方程:
p/[V(po-p)]=1/(VmC)+(C-1)p/(VmCpo)
(2)
C≈exp[(Q1-QL)/(RT)]
(3)
式中:V為單位質(zhì)量樣品表面氮氣吸附量;Vm為單位質(zhì)量樣品表面單分子層氮氣飽和吸附量;po為在液氮溫度下氮氣的飽和蒸氣壓;p為測定吸附量時氮氣分壓;C為與材料吸附特性相關(guān)的常數(shù),由吸附儀直接計算給出;Q1為第一層的吸附熱;QL為吸附質(zhì)的液化熱。
根據(jù)C的物理意義可知,C值越大表示固體材料內(nèi)表面吸附能力越強(qiáng),吸附層數(shù)越多。若C>1,表示吸附劑與吸附質(zhì)分子之間的吸引力大于吸附質(zhì)為液體時分子之間的引力[6]。
根據(jù)多次對比試驗,添加不同濃度的MEA、DEA、TEA溶液為堿性催化劑,可得到不同效果的硅氣凝膠。試驗選擇乙醇胺水溶液濃度分別為:MEA,1∶20;DEA,1∶5;TEA,1∶5(均為體積比)。
分別將MEA、DEA、TEA溶液和氨水按照不同的加入量加入到TEOS水解液中,凝膠置換溶劑后不經(jīng)表面改性,采用真空干燥得到SiO2氣凝膠。測試比表面積,得到相應(yīng)的C值。堿催化劑添加量與C值的關(guān)系見表1。由表1可知:當(dāng)堿催化劑添加量為7.6×10-3mol時,TEA的C值最大,MEA的C值次之,約為TEA的C值的1/2;當(dāng)堿性催化劑添加量增加到8.5×10-2mol時,TEA的C值降到最低值;對于MEA和DEA,當(dāng)其添加量由7.6×10-3mol增加到1.7×10-2mol時,二者的C值降到最低,隨后隨著添加量的增加C值上升;氨水的C值隨著氨水添加量的增加呈下降趨勢。從表1還可以看出,MEA在添加量為(5.1~8.5)×10-2mol的較寬范圍內(nèi),其C值比DEA、TEA和氨水的C值高,但在低添加量處TEA的效果最好,MEA和DEA的效果次之。說明MEA作為堿性催化劑的加入,有助于使氣凝膠內(nèi)表面的吸附能力增強(qiáng),使吸附量增大,吸附更多層的分子,MEA的作用效果要優(yōu)于DEA和氨水。該特性使氣凝膠可能對于某些極性分子的吸附能力增強(qiáng),有助于提高選擇性吸附和吸附量。
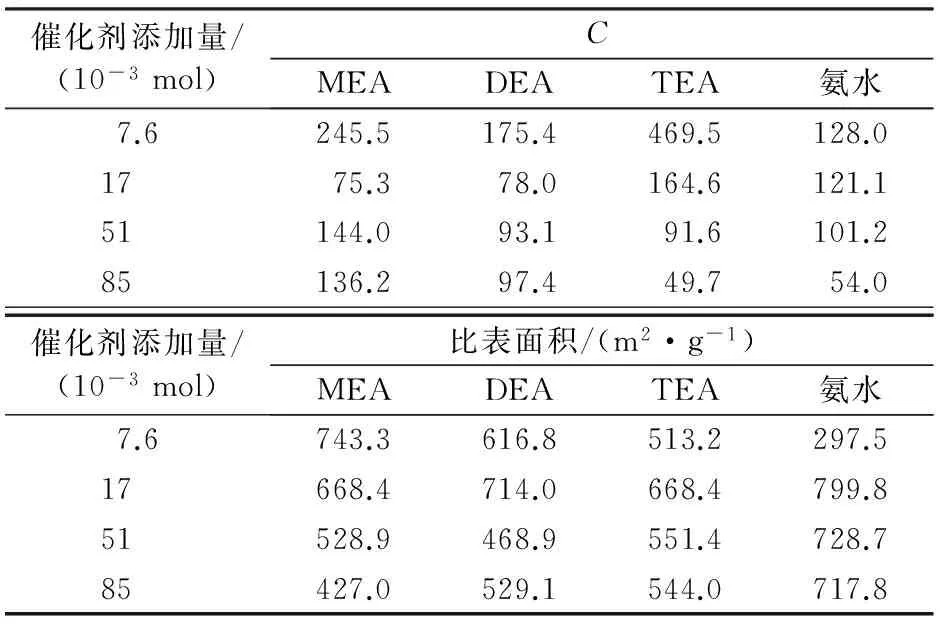
表1 堿性催化劑添加量與硅氣凝膠吸附參數(shù)C、比表面積的關(guān)系
由表1看出:在堿性催化劑低加入量處(7.6×10-3mol),以MEA為催化劑所得氣凝膠的比表面積最大,為743.3 m2/g,是以氨水為催化劑的2.5倍,并且隨著MEA添加量的增加,氣凝膠比表面積逐步下降;而DEA和TEA具有相似的規(guī)律,在添加量為1.7×10-2mol處,氣凝膠比表面積最大,然后隨著催化劑添加量的增加,氣凝膠比表面積下降;以氨水為催化劑的變化趨勢與前面不同,在添加量為7.6×10-3mol處氣凝膠比表面積最小,在添加量為1.7×10-2mol時氣凝膠比表面積最大,然后隨著添加量的增加比表面積逐步下降,但下降幅度不大。究其原因主要是,在相同添加量下乙醇胺水溶液的堿性高于氨水,堿性過強(qiáng)易使局部形成的凝膠顆粒快速縮聚,形成的骨架顆粒粗大,孔徑較大,進(jìn)而使比表面積減小。
2.2 N2吸附-脫附等溫曲線的特征
圖1為MEA、DEA、TEA和氨水添加量為7.6×10-3mol時所得SiO2氣凝膠典型N2吸附-脫附等溫線。從圖1看出,DEA為催化劑的吸附等溫線為I型等溫線,其余的吸附等溫線為II型等溫線。I型等溫線是單分子吸附層曲線,隨著相對壓力的不斷提高,發(fā)生毛細(xì)管凝聚,使吸附量增加,一旦將所有的微孔填滿后,吸附量便不會隨著相對壓力的提高而增加,呈現(xiàn)出吸附飽和。對于以MEA、TEA和氨水溶液為催化劑的II型吸附等溫線,吸附分支變化緩慢,而脫附分支在中等大小相對壓力處有一陡的變化,具有細(xì)頸和墨水瓶形狀的孔可以有此類吸附回線。在吸附分支上,當(dāng)相對壓力增至與瓶頸半徑相對應(yīng)的值時,凝聚液便開始充滿瓶頸,隨著相對壓力繼續(xù)增加,連續(xù)地充滿整個墨水瓶體。所以,吸附分支是逐漸變化的。在脫附分支上,由于瓶頸上的液體將廣體中的液體封住了,一直到相對壓力降至與瓶頸對應(yīng)的值時便發(fā)生驟然的脫附至空[6]。比較乙醇胺硅氣凝膠的吸附-脫附曲線,在相對壓力大于0.53時TEA的吸附線要高出MEA、DEA和氨水較多,說明其吸附能力要高于MEA和DEA,這與TEA的C值較MEA和DEA的C值高有相同的規(guī)律。而在相對壓力低于0.53時,氨水的吸附-脫附曲線稍高于其他吸附-脫附曲線。從圖1還可以看出,TEA、MEA和氨水為催化劑硅氣凝膠孔徑分布有一定的范圍,而DEA為催化劑的硅氣凝膠孔徑分布較為均勻。
圖2為MEA、DEA、TEA、氨水添加量為7.6×10-3mol所得SiO2氣凝膠吸附孔徑分布圖。由圖2可知,以氨水和TEA為堿性催化劑時所得二氧化硅氣凝膠孔徑最小,最高峰位于1.7 nm;DEA為1.93 nm;MEA最大,為2.18 nm,是氨水的1.28倍。這說明,MEA能夠形成較大的孔徑,孔徑的適當(dāng)增大有利于降低溶劑揮發(fā)時的界面張力,減少了孔的收縮和坍塌。從孔徑分布的幅度來看,TEA波峰的寬度最大,而氨水的最小,說明以TEA為堿性催化劑所得的氣凝膠孔徑分布范圍較廣,這主要與TEA多羥基和SiO2表面羥基形成氫鍵影響了孔結(jié)構(gòu)有關(guān),而氨水的則比較單一。DEA也可得到孔徑分布較窄的硅氣凝膠。
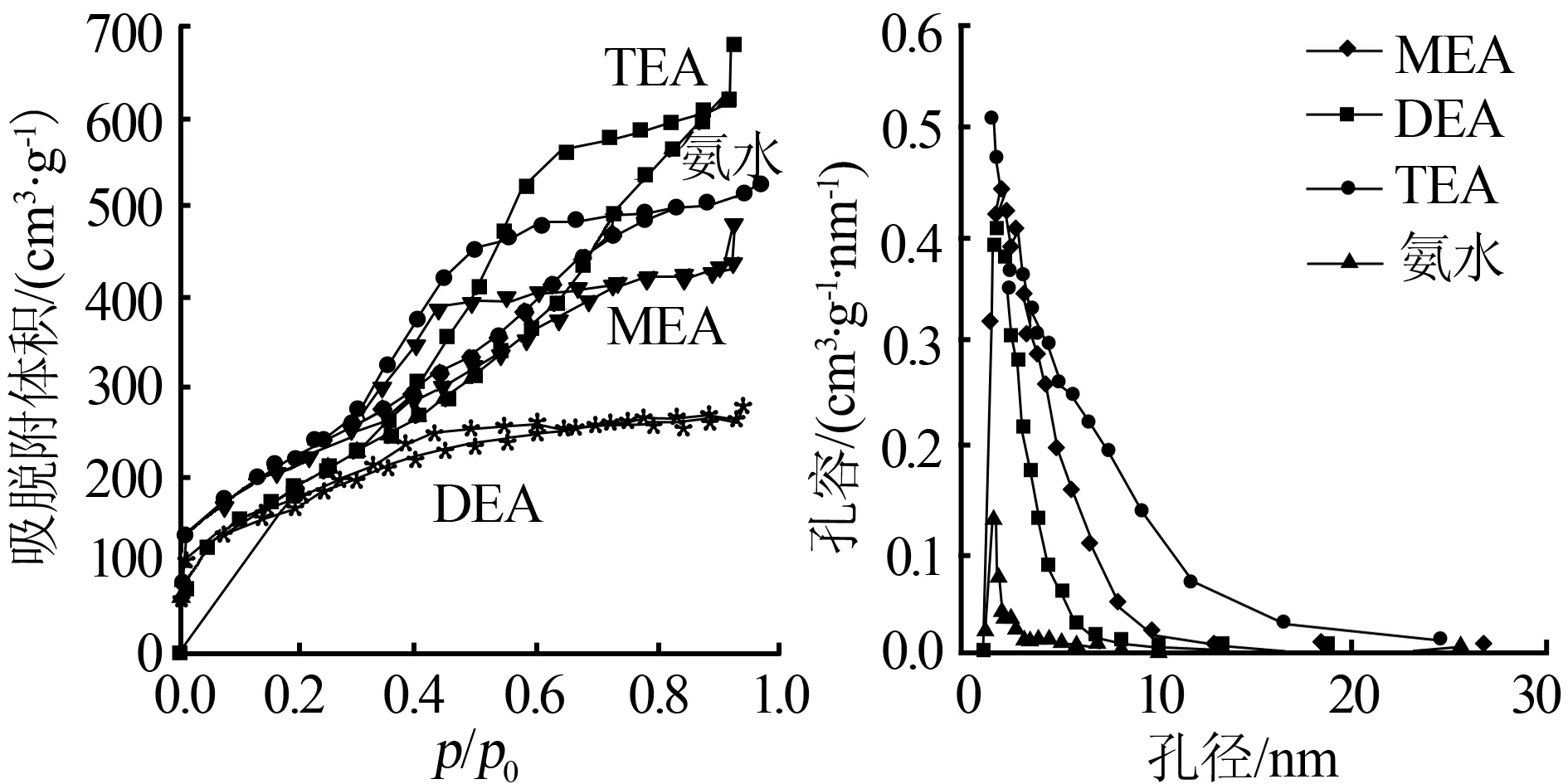
圖1不同堿性催化劑的吸脫附曲線圖2不同堿催化劑的孔徑分布圖
表2為堿性催化劑添加量與硅氣凝膠平均孔徑、孔隙率的關(guān)系。由表2看出:隨著MEA添加量的增加,孔徑的變化呈拋物線狀,在添加量為5.1×10-2mol時出現(xiàn)最大值,添加量再增大則孔徑下降幅度較大;隨著TEA添加量的增加,孔徑呈逐漸增大趨勢,當(dāng)添加量達(dá)到5.1×10-2mol之后,孔徑增大不明顯,其最大值低于MEA;氨水和DEA的變化規(guī)律相似,隨著添加量的增加,平均孔徑增加。究其原因主要是MEA溶液堿性過強(qiáng),添加量超過一定量,易使凝膠顆粒發(fā)生粘連,使孔徑變小。從表2看出:隨著MEA、DEA添加量的增加,孔隙率呈增加的趨勢,但添加量超過5.1×10-2mol后孔隙率增加不明顯;而氨水則在其添加量為1.7×10-2mol 后,孔隙率有較大的上升幅度;隨著TEA添加量的增加,孔隙率在出現(xiàn)峰值后則出現(xiàn)下降趨勢。表2數(shù)據(jù)說明,以乙醇胺作為催化劑,在低添加量時所得硅氣凝膠的孔隙率高于氨水作催化劑的孔隙率。
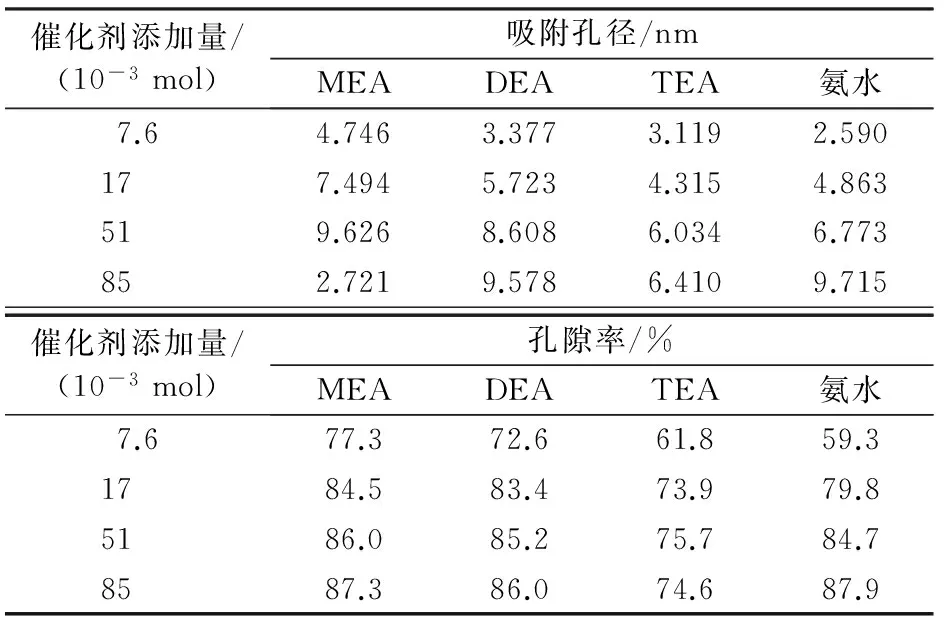
表2 堿催化劑添加量與硅氣凝膠吸附孔徑、孔隙率的關(guān)系
2.3 表面形貌和微觀結(jié)構(gòu)
圖3為分別加入MEA、DEA和TEA所得SiO2氣凝膠的掃描電鏡照片,MEA添加量為7.6×10-3mol,DEA、TEA添加量為1.7×10-2mol。從圖3看出,所用堿性催化劑不同,SiO2氣凝膠的微觀結(jié)構(gòu)不同。圖3a中SiO2呈顆粒狀,幾個顆粒互相粘連形成小團(tuán),團(tuán)與團(tuán)之間鱗次櫛比,中間有小孔隙存在。圖3b中SiO2部分呈現(xiàn)正四面體顆粒狀,其余則是很小的顆粒,形成網(wǎng)狀結(jié)構(gòu)。圖3c與圖3a樣品形狀相似,但是出現(xiàn)較多的“粘連”,形成團(tuán)聚現(xiàn)象,使其比表面積、孔隙率下降。分析其原因可能是由于TEA具有3個羥基,氫與硅氣凝膠表面羥基中的氧形成氫鍵,從而使一分子的TEA可“拉住”3個凝膠顆粒,發(fā)展開來,則更易形成粘連現(xiàn)象。而MEA和DEA羥基少,形成的凝膠顆粒較分散。

圖3 SiO2氣凝膠SEM照片
2.4 表面特性
通過N2吸附等溫線可檢測出不同堿性催化劑對SiO2氣凝膠內(nèi)部表面不規(guī)則結(jié)構(gòu)形狀的影響。將不同添加量下所得樣品的p0/p取對數(shù)后,與V/Vm作圖得到直線,計算出斜率m,根據(jù)文獻(xiàn)[7],斜率表示了吸收毛細(xì)管冷凝的機(jī)制。根據(jù)公式Ds=3-|1/m|計算出Ds(表面不規(guī)則數(shù)),Ds越大表明其內(nèi)表面越粗糙。將不同堿催化劑添加量下的Ds作圖,見圖4。由圖4看出,TEA的Ds除了在第一個點低于氨水,其余的點始終在其他線的上方,說明以TEA為堿性催化劑得到的硅氣凝膠相對于其他堿催化劑其內(nèi)部較粗糙。
圖5為添加不同堿性催化劑制備的SiO2氣凝膠的紅外吸收光譜。其中460、800[8]、1 080、1 650 cm-1[9]附近的吸收峰是Si—O—Si的振動吸收峰;3 466 cm-1附近的吸收峰代表O—H的吸收帶,是物理吸附水產(chǎn)生的,這些峰是SiO2氣凝膠的典型峰。960 cm-1是Si—OH 的伸縮振動,二乙醇胺的峰值較弱,可認(rèn)為該樣品中具有較少的羥基基團(tuán)。2 970 cm-1和930 cm-1[10]附近的吸收峰為—CH3的不對稱振動和搖擺振動,說明在表面出現(xiàn)了硅甲基,圖5中MEA在該處的振動強(qiáng)度明顯增強(qiáng),而DEA的則相對弱很多。對于氨水,在2 970 cm-1處無峰出現(xiàn)。該峰常出現(xiàn)于使用三甲基氯硅烷(TMCS)表面改性劑進(jìn)行表面處理的硅氣凝膠,即TMCS中的—C2H5基團(tuán)掛在了濕凝膠的表面[11]。但本研究中并未使用TMCS等表面改性劑,那么這個峰可能是Si—O—C2H5的對稱伸縮振動,這也導(dǎo)致了譜圖在2 970 cm-1處出現(xiàn)了C—H鍵的對稱和不對稱伸縮振動特征峰[12],該峰還顯示了SiO2氣凝膠具有一定的疏水性能。—O—C2H5基團(tuán)只能來自于TEOS中,MEA能促使TEOS的水解反應(yīng)朝逆向進(jìn)行,增強(qiáng)了硅氣凝膠的疏水性。
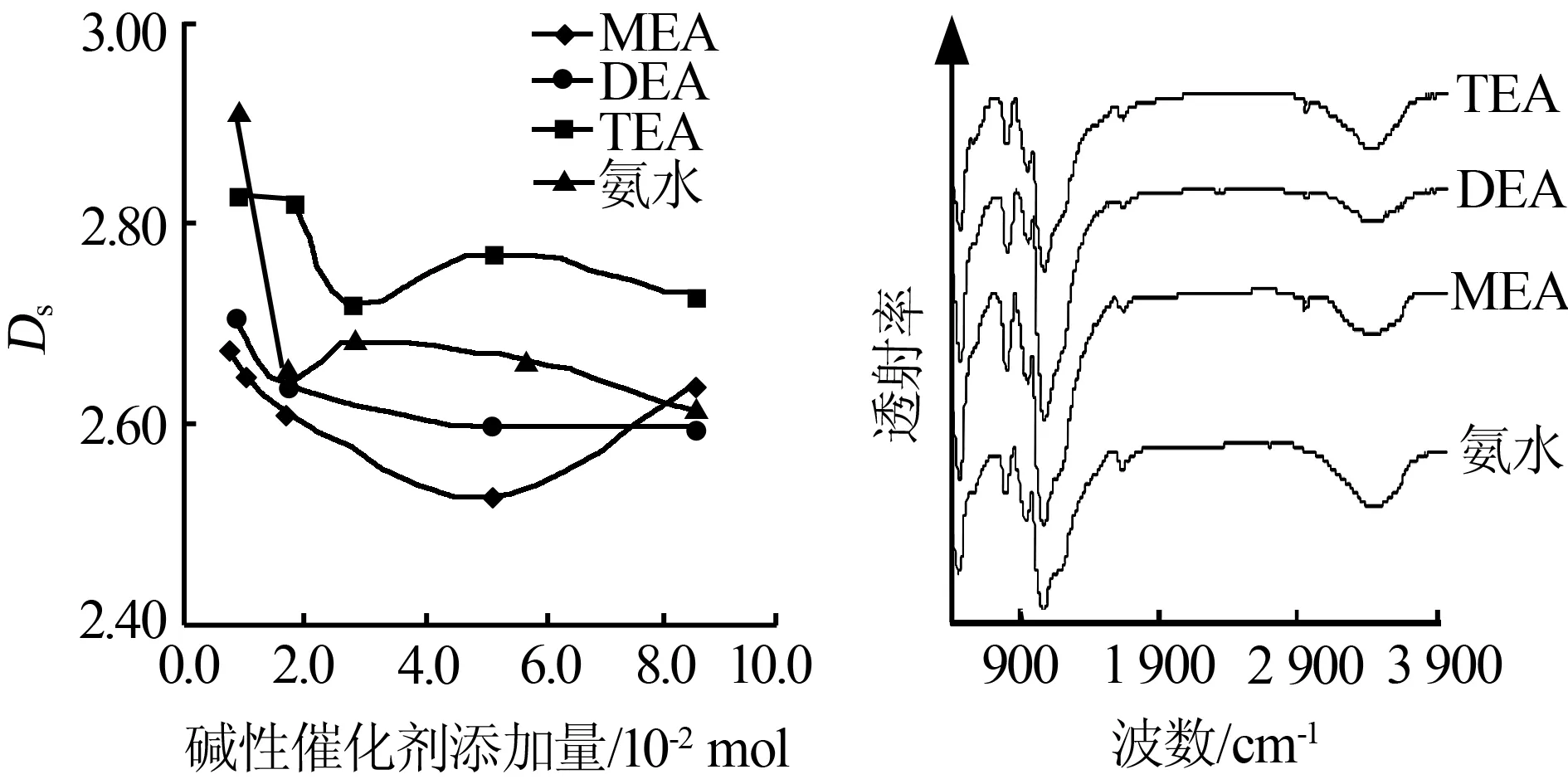
圖4堿催化劑添加量與Ds關(guān)系圖圖5不同堿催化劑制備SiO2氣凝膠紅外光譜圖
3 結(jié)論
1)以TEOS為硅源,采用酸/堿催化法,使用不同體積比乙醇胺溶液作為堿性催化劑,在不同添加量下,可得到孔隙率為61.8%~87.3%、平均孔徑為2.7~9.6 nm的硅氣凝膠。在低添加量時,乙醇胺作催化劑所得硅氣凝膠的孔隙率高于氨水,平均孔徑大于氨水;2) 使用TEA作堿性催化劑,在添加量為7.6×10-3mol時,其C值明顯高于氨水的C值,可提高N2吸附量;3)加入TEA所得SiO2氣凝膠,其內(nèi)表面較加入氨水的粗糙,使吸附能力增強(qiáng);4)使用MEA作堿性催化劑,可使SiO2氣凝膠具有一定的疏水性。
[1] 張偉娜,李云輝,王慶偉,等.二氧化硅氣凝膠的表面改性及熱穩(wěn)定性的研究[J].吉林師范大學(xué)學(xué)報:自然科學(xué)版,2009,37(2):67-69.
[2] Woignier T,Phalippou J,Vacher R.Parameters affecting elastic properties of silica aerogels[J].J.Mater.Res.,1989,4(3):668-692.
[3] Yoda S,Ohshima S,Kamiya K.et al.Effects of ethanolamines catalysts on properties and microstructures of silica aerogels[J].J.Non-Cryst.Solids,1996,208(1/2):191-198.
[4] Carre F,Cerveau G,Chuit C,et al.Hexacoordination at silicon: the case of silatranes[J].Organometallics,1990,9(7):1989-1991.
[5] Nakanishi K,Minakuchi H,Soga N,et al.Structure design of double-pore silica and its application to HPLC[J].J.Sol-Gel Sci.Technol.,1998,13(1/2/3):163-169.
[6] 沈鐘,王果庭.膠體與表面化學(xué)[M].北京:化學(xué)工業(yè)出版社,2004:147-308.
[7] Pfeifer P,Cole M W.Fractals in surface science:scattering and thermodynamics of adsorbed films II[J].New J.Chem.,1990,14(3):221-232.
[8] 何方,趙紅雨,徐三魁.硅石氣凝膠老化的新方法[J].河南化工,2008,25(8):19-20.
[9] 史非,王立久,劉敬肖,等.介孔SiO2氣凝膠的常壓干燥制備研究[J].無機(jī)化學(xué)學(xué)報,2005,21(11):1632-1636.
[10] 張志華,倪星元,沈軍,等.SiO2氣凝膠表面特性對其吸附性能的影響[J].材料導(dǎo)報,2005,19(7):115-117.
[11] 王娟,張長瑞,馮堅.三甲基氯硅烷對納米多孔二氧化硅薄膜的修飾[J].物理化學(xué)學(xué)報,2004,20(12):1399-1403.
[12] 吳國友,余煜璽,程璇,等.甲基三甲氧基硅烷對塊狀SiO2氣凝膠性能和結(jié)構(gòu)的影響[J].硅酸鹽學(xué)報,2009,37(7):1206-1211.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學(xué)學(xué)報(工學(xué)版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(xué)(2015年4期)2016-01-17 09:01:27
應(yīng)用化工(2014年3期)2014-08-16 13:23:50
