柱前衍生化-HPLC檢測丙戊酸鈉血清濃度方法驗證
2010-07-12 10:35:56曲秀梅雷力力張志國
黑龍江醫藥科學 2010年5期
曲秀梅,雷力力,方 芳,李 冬,張志國
(佳木斯大學附屬第一醫院,黑龍江 佳木斯 154003)
丙戊酸鈉為一線廣譜抗癲癇藥物,因分子結構中沒有能夠產生紫外光譜吸收的結構,用一般的 HPLC法難于直接測定,文獻報道多用毛細管氣相色譜法、酶聯免疫法、熒光偏振免疫法等。目前應用較多的是“柱前衍生化高效液相色譜法檢測丙戊酸鈉的血清含量”,為了開展丙戊酸鈉臨床血藥濃度監測,對文獻[1,2]報道的柱前衍生化-高效液相色譜檢測方法進行了綜合,經過改進的方法檢測結果穩定、準確,可以投入臨床使用。
1 儀器及試劑
P230Ⅱ高效液相色譜儀(大連伊利特分析儀器儀器有限公司);XW-80渦旋混合器(上海精科實業有限公司);島津AUW220D十萬分之一天平。
色譜純甲醇:天津凱通化學試劑有限公司生產;丙戊酸鈉對照品 (湖南省湘中制藥有限公司提供,含量:99.8%,批號 090405)環己烷羧酸 (美國 Sigma公司 ,批號:08265PD,);ω-溴苯乙酮(國藥集團化學試劑有限公司,批號:20090212);其它試劑均為分析純。
2 方法及結果
2.1 色譜條件
色譜柱:Hypersil ODS2C18(4.6mm× 250mm,5μm);柱溫:30℃;檢測波長:248nm;流動相:甲醇:水 (75:25);流速:1.0mL˙min-1;進樣量:15 μL。
2.2 試驗溶液的制備
精密稱取丙戊酸鈉對照品37.6mg,置50mL容量瓶中,用 0.2mol˙L-1氨水稀釋至刻度 (含丙戊酸鈉 0.752mg˙ mL-1),搖勻,作為丙戊酸鈉標準溶液。精密稱取 50.2mg環己烷羧酸,置50mL容量瓶中 ,用 0.2mol˙L-1氨水溶解并稀釋至刻度,再精密量取該液5mL,置 25mL容量瓶中,0.2mol˙L-1氨水稀釋至刻度,搖勻,作為內標溶液(含環己烷羧酸 0.2008mg˙mL-1)。精密稱取125.4mg ω-溴苯乙酮,置25mL容量瓶中,用乙腈溶解并稀釋至刻度,搖勻 ,作為衍生化試液 (含 ω-溴苯乙酮 5.016mg˙mL-1),放置于冰箱中保存。
2.3 樣品制備及衍生化
取 200μL血清置空白采血管中,加入 30μL內標溶液和20 μL丙戊酸鈉標準溶液 ,再加入 200 μL 1mol˙L-1的硫酸溶液,振搖混勻,然后加入4mL正己烷振搖萃取3min,靜置5min使充分分層,分離出上清液2mL于另一試管中,加入40μL衍生化試液和20μL三乙胺,漩渦混勻,于63℃水浴衍生 10min,然后在氮氣流下蒸發至干,立即取出,用甲醇0.3mL漩渦溶解 ,作為血清樣品溶液。
2.4 色譜行為
在本實驗條件下,衍生試劑峰與丙戊酸鈉及內標衍生物能很好地分離,血清中無明顯干擾峰。內標衍生物保留時間為7.00min,丙戊酸鈉衍生物保留時間為10.90min。色譜圖見圖1。
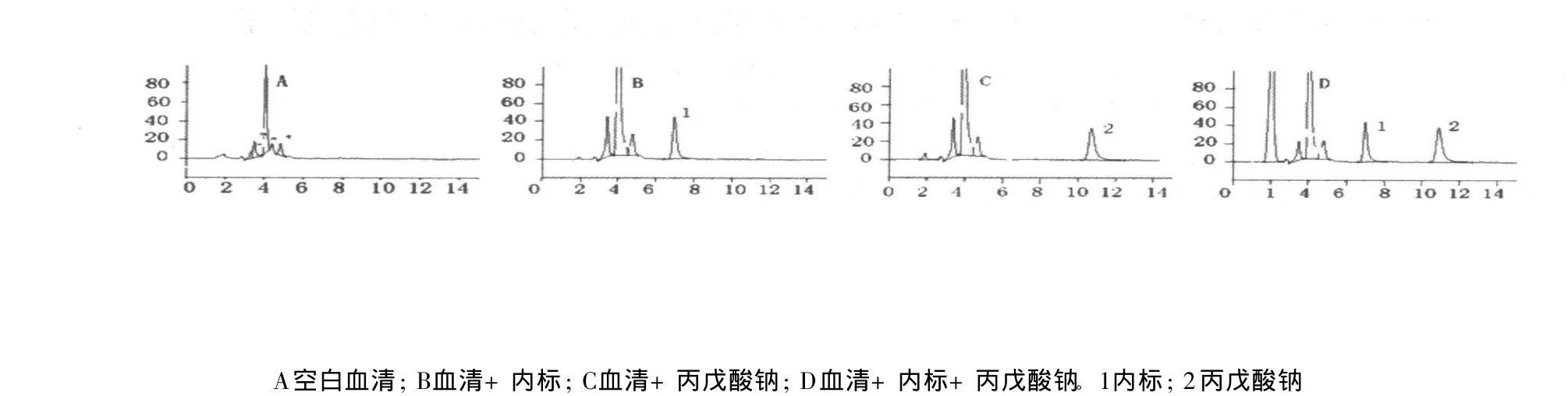
圖1 HPLC色譜圖
2.5 標準曲線的制作
按 2.3項下方法 ,分別制備 8份樣品溶液 ,丙戊酸鈉標準溶 液 加入 量 分別為:18.80、37.60、56.40、75.20、94.00、112.80、127.84、150.40 μg˙mL-1,按色譜條件分別進樣 ,計算各自的標準濃度(Y)對其峰高與內標峰高比值(X)的線性回歸,其線性關系式為 Y= 86.852X+ 1.9179,r= 0.9988,線 性 范 圍 為 18.80μ g~ 150.40μg˙mL-1,定 量 下 限 為18.80 μg˙mL-1。
2.6 精密度及回收率試驗
按 2.3項下方法,分別制備高中低三個濃度 (94.00、75.20、56.40 μg˙mL-1)血清樣品溶液各 5份 ,連續測定 3d,以內標法計算其方法回收率及日內、日間精密度。結果見表1。
表1 精密度及回收率試驗結果 ±s)

表1 精密度及回收率試驗結果 ±s)
日內精密度(n=5)日間精密度(n=15)加入量μ g˙mL-1 檢出量μ g˙mL-1 回收率(%)RSD%檢出量μ g˙mL-1 回收率(%)RSD%94.00 89.99±2.41 95.73 2.68 93.25±0.38 99.51 8.49 75.20 75.02±7.05 99.76 9.40 73.79±3.43 98.13 4.65 56.40 56.24±1.37 99.72 2.44 57.65±3.39 102.22 5.89
2.7 穩定性試驗
2.7.1 衍生物穩定性試驗
按 2.3制備高、中、低三個濃度 (94.00、75.20、56.40 μg˙ mL-1)的樣品溶液,分別于 0、24、48h時各進樣 2次,以丙戊酸鈉與內標峰面積比值計算 RSD,各樣品 RSD為:0.82%;0.82%;1.30%。說明樣品在48h內檢測,結果穩定。
2.7.2 含藥血清樣品穩定性試驗
取 血 清 9mL ,分 為 高、中、低 (94.00、75.20、56.40 μg˙mL-1)3個濃度組,各組分別加入丙戊酸鈉標準溶液 375μL、300μL和 225μL,漩渦混勻 ,置冰箱冷藏 (2~ 10℃ )保存 ,然后于 0、5、10d分別取血清 200 μL各 5份 ,各加入 30μL內標溶液 ,從加入 200μL1mol˙L-1的硫酸溶液開始 ,按 2.3項下方法制備樣品溶液。取水200 μL,從加入30μL內標溶液開始按2.3項下方法制備對照品溶液。按2.1色譜條件分別檢測對照品溶液和樣品溶液,記錄色譜圖。以內標法計算各樣品回收率和各濃度回收率的 RSD。結果,高、中、低三個濃度樣品回收率和 RSD分別為95.44±7.65,RSD=8.02%;93.92±3.37,RSD=3.59%;99.32±3.81,RSD=3.83%;說 明血清樣品置冰箱(2~ 10℃)冷藏保存,10d內檢測,結果穩定。
3 討論
3.1 檢測波長的確定
文獻[1~4]報道檢測波長有 266、248、235和 254nm,本文作者在以甲醇為溶劑,丙戊酸鈉含量為0.752mg˙mL-1于200nm~280nm進行紫外掃描,上述各個波長處均有吸收,當濃度減少為0.2mg˙mL-1時,266nm波長處吸收很小 ,其他波長有吸收,隨著波長減小,吸收度加大;當以同一濃度(0.752mg˙mL-1)樣品 ,分別在 266、248、235和 254nm檢測 ,丙戊酸鈉及環己烷羧酸峰高按248、254、235和266nm依次遞減,因而本文選擇248nm作為吸收波長進行試驗。
3.2 提取衍生溶媒的選擇
文獻報道提取衍生化溶媒有二氯甲烷[1]和正己烷[2],本文在用二氯甲烷作為溶媒,在248nm檢測時,空白溶液在內標和丙戊酸鈉保留時間處有吸收峰出現,改變流動相比例不能使雜質峰分開;在266nm處,空白、內標、丙戊酸鈉和樣品溶液均沒有吸收峰出現,可能是檢測濃度較低所致。因而選用正己烷作為衍生化溶媒。
3.3 蒸干終點的確定
在接近蒸干時,應注意隨時觀察,發現已蒸干立即取出,否則將會出現環己烷羧酸峰減小,丙戊酸鈉峰加大現象。
3.4 計算方法的確定
內標法計算血清樣品濃度,可以用峰高或峰面積[5]計算,試驗結果顯示以峰高計算樣品濃度較以峰面積計算樣品濃度準確,穩定,因而采用峰面積計算樣品含量。
3.5 衍生化試劑用量的確定
有報道[6]衍生化試劑用量為2.5~ 10倍,本試驗用量為7.33倍,當小于7.33倍時,線性關系試驗中的高濃度部分會出現非理想峰高和峰形,因而選用本實驗用量。
[1]孫志宏,陽國平,裴奇,等.2種丙戊酸鈉片劑在人體內的相對生物利用度研究 [J].中南藥學,2009,7(6):415
[2]陳堅,方維軍,吳芳,等.高效液相色譜法測定丙戊酸血藥濃度[J].中國臨床藥學雜志,2000,9(5):296
[3]展翔,徐枚.高效液相色譜法測定人血清中丙戊酸鈉含量 [J].中國藥業,2009,18(6):25
[4]石蘇英,沈建幸.快速柱前衍生-高效液相色譜法測定丙戊酸血藥濃度 [J].中國現代應用藥學雜志,2007,24(4):315
[5]國家藥典委員會編.中華人民共和國藥典 [M].2005年版二部.北京:化學工業出版社,2005,28
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中老年保健(2021年3期)2021-08-22 06:50:04
昆明醫科大學學報(2021年1期)2021-02-07 01:06:36
現代臨床醫學(2021年1期)2021-01-26 00:56:02
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中華養生保健(2020年4期)2020-11-16 01:31:40
中西醫結合肝病雜志(2020年2期)2020-10-27 02:18:50
