緊密連接相關蛋白Claudin-5、ZO-1在大鼠蛛網膜下腔出血后早期腦損傷中的表達及意義
2010-05-25 01:43:28陳鐸袁江偉宋磊魏翔泰關俊宏劉云會宗志紅
中國醫科大學學報 2010年9期
陳鐸,袁江偉,宋磊,魏翔泰,關俊宏,劉云會,宗志紅
(中國醫科大學 1.附屬盛京醫院神經外科,沈陽 110004;2.基礎醫學院生化教研室,沈陽 110001)
最新的研究結果顯示,蛛網膜下腔出血(subarachnoid hemorrhage,SAH)后72 h內存在早期腦損傷(early brain injury,EBI),3~5 d 后發生腦血管痙攣(cerebral vasospasm,CVS),上述二者是造成SAH患者死亡或重殘的最主要原因[1]。然而,EBI的確切分子病理機制尚不清楚,細胞凋亡機制可能參與了其發生、發展過程[2]。c-Jun 氨基端激酶(c-Jun N terminal kinase,JNK)信號轉導通路參與了多種疾病的細胞凋亡過程[3]。腦損傷后發生的腦水腫病理改變與血腦屏障的開放密切相關,Claudin-5和ZO-1是最重要的腦血管內皮細胞緊密連接相關蛋白,它們的異常在血腦屏障開放中起關鍵作用[4,5]。本研究擬通過檢測大鼠SAH后早期腦皮層微血管緊密連接相關蛋白Claudin-5和ZO-1的表達及JNK抑制劑對其表達的影響,進一步探討SAH后早期腦損傷的機制。
1 材料與方法
1.1 實驗動物及分組
健康成年雄性SD大鼠75只,體質量300~350 g,由中國醫科大學附屬盛京醫院實驗動物中心提供。采用隨機數字表法將大鼠分為假手術組(sham組)、蛛網膜下腔出血(SAH)組、SAH+DMSO組、SAH+SP600125(10 mg/kg)組 和 SAH+SP600125(30 mg/kg)組,每組15只。SAH+二甲基亞砜(dimethyl sulfoxide,DMSO)、SAH+SP600125(10 mg/kg)和SAH+SP600125(30 mg/kg)組。在建立SAH模型基礎上分別于實驗前1h和實驗后6h向腹腔內二次注入載體藥物DMSO(Calbiochem公司)或JNK抑制劑SP600125(Sigma公司),每組大鼠中5只用于電鏡形態學觀察,10只用于Western blot檢測Claudin-5、ZO-1的表達。
1.2 大鼠SAH模型的建立
采用血管內穿刺法建立大鼠蛛網膜下腔出血模型[6]:用 10%水合氯醛(300 mg/kg)經腹腔注射麻醉后,氣管內插管,固定頭部及四肢,做頸部正中3 cm切口,分離暴露右側頸總動脈、頸內動脈、頸外動脈,電凝切斷頸外動脈近分叉處分支(包括枕動脈、甲狀腺上動脈及咽升動脈),分離結扎并切斷頸外動脈,使頸外動脈近心端成一約5 mm長的殘端,用2個無損傷動脈夾分別夾閉頸總動脈和頸內動脈,在頸外動脈起始段約3~5 mm處用眼科剪剪一“V”型小切口,下拉頸外動脈,與頸內動脈成一直線,再將1根長約5 cm的3-0尼龍線從頸外動脈上的“V”型小切口導入,經頸總動脈達頸內動脈,松開頸內動脈上的動脈夾,順著頸內動脈向遠端繼續插入尼龍線至頸內動脈顱內段,有小的阻力感后再插入約2~3 mm刺破頸內動脈分叉部,迅速抽出尼龍線,用動脈夾夾住頸外動脈殘端,松開頸總動脈上的動脈夾恢復血流,結扎頸外動脈殘端,縫合頸部切口。術中股動脈插管接多導生理儀監測血壓,維持肛溫(37.5±0.5)℃,并定時測血氣分析。sham組除了不刺破血管壁外,其余操作均與SAH組一致。
1.3 透射電鏡觀察大鼠腦皮層微血管超微結構
SAH模型建立24 h后處死大鼠,2%戊二醛溶液進行全身灌流,在冰盤內摘取大鼠腦皮質組織,迅速切成1 cm×1 cm×1 cm的小塊,3%戊二醛溶液中預固定,0.1 mol/L磷酸緩沖液漂洗,1%四氧化鋨固定,梯度乙醇脫水,定向包埋,定位后制成超薄切片,經醋酸雙氧鈾-枸櫞酸鉛雙染色后,于透射電鏡下觀察、鑒別皮層神經元與皮層微血管組織,并照像。觀察腦皮層微血管超微結構變化。
1.4 Western blot檢測Claudin-5、ZO-1表達的變化
取各組大鼠新鮮大腦皮質,常規提取蛋白,10%SDS聚丙烯酰胺凝膠電泳分離蛋白樣品,4℃轉膜過夜,5%脫脂奶粉封閉;加入1∶500稀釋山羊抗鼠多克隆抗體(Santa公司),4℃孵育過夜;二抗室溫孵育2 h,DAB顯色,照像。以GAPDH作為內對照。膠片掃描后,用Scion Image for Windows軟件分析各條帶的平均灰度值。
1.5 統計學分析
應用SPSS13.0統計軟件。計數資料用率表示,采用t檢驗;計量資料用x±s表示,各組之間行ANOVA方差分析。P<0.05表示有統計學差異。
2 結果
2.1 大鼠SAH后24 h神經行為功能缺損評分
SAH后24 h對存活大鼠參照Bederson法[7]進行神經行為功能缺損評分,結果見表1。在SAH組和SAH+DMSO組中神經行為功能缺損評分集中在3 分和 4 分;SAH+SP600125(10 mg/kg)組集中在 2分和3分,而SAH+SP600125(30 mg/kg)組集中在1分和2分,說明JNK抑制劑SP600125可改善SAH存活大鼠神經行為功能缺損,SP600125(30 mg/kg)組尤為明顯;sham組無死亡,SAH組、SAH+DMSO組、SAH+SP600125(10 mg/kg) 組、SAH+SP600125(30 mg/kg)組死亡率分別為30%、35%、25%、15%,各組間比較無統計學差異(P>0.05)。
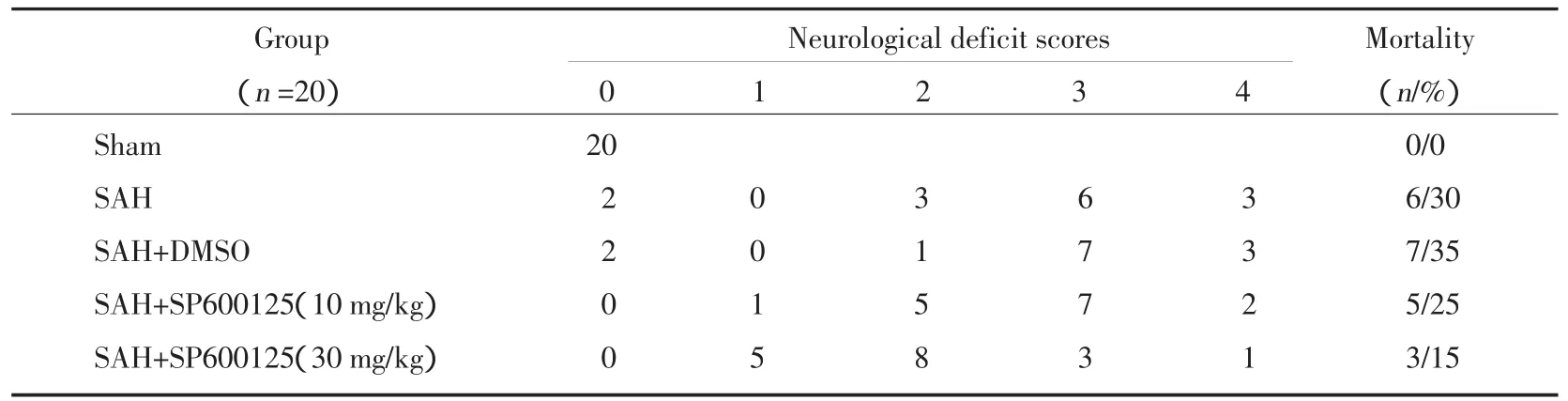
表1 SAH后24 h大鼠神經行為功能缺損評分及死亡率Tab.1 Neurological deficit scores and mortality after SAH
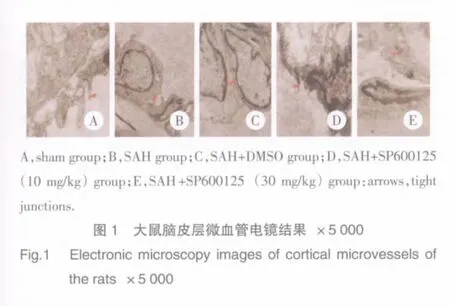
2.2 SAH大鼠腦皮層微血管組織形態學變化及SP600125抑制劑對其影響
透射電鏡的結果(圖1)顯示,sham組大鼠腦皮層微血管內皮細胞呈梭形,胞膜連續完整,細胞核形狀不規則,核膜尚完整,胞質內有較多核糖體、線粒體及粗面內質網等細胞器,相鄰內皮細胞間可見緊密連接,血腦屏障未開放;SAH組和SAH+DMSO組血管內皮細胞多凸向管腔,細胞核形不規則,有切跡,核內異染色質明顯邊集,細胞基質密度降低,線粒體等細胞器減少,相鄰內皮細胞間緊密連接明顯松散,血腦屏障明顯開放;SAH+SP600125(10 mg/kg)組細胞核形不規則,有切跡,核內異染色質明顯邊集,核膜部分模糊,細胞質內囊泡增多,相鄰內皮細胞間緊密連接近腔面有部分松散,血腦屏障呈部分開放;SAH+SP600125(30 mg/kg)組細胞核形不規則,有切跡,細胞膜部分破壞,胞質內可見空泡,變性的線粒體、粗面內質網擴張,相鄰內皮細胞間緊密連接呈部分松散狀態,血腦屏障呈部分開放。
2.3 SAH大鼠腦皮層微血管緊密連接相關蛋白Claudin-5和ZO-1的表達及SP600125對其影響
Western blot結果(圖2)表明,SAH組和 SAH+DMSO組Claudin-5和ZO-1表達水平下調,較sham組具有統計學差異(P<0.05);SAH組和 SAH+DMSO組之間比較,無統計學差異;與SAH、SAH+DMSO 組比較,SAH+SP600125(10 mg/kg)組Claudin-5和ZO-1的表達下調未受到明顯抑制(P>0.05);SAH+SP600125(30 mg/kg)組 Claudin-5 和 ZO-1 的表達下調則受到明顯抑制,10 mg/kg與30mg/kg SP600125兩組間比較具有統計學差異(P<0.05)。
3 討論
3.1 SAH后早期腦損傷

自發性蛛網膜下腔出血后早期腦損傷是指SAH后72 h內發生的一系列腦組織損害[8]。長期以來眾多學者研究的重點多集中在SAH后3~5 d延遲發生的腦血管痙攣機制的研究,我們的前期研究結果也證實了遲發性腦血管痙攣是影響預后的關鍵因素之一[9,10]。然而,最近的研究結果提示,EBI也是造成患者死亡或重殘的最主要原因之一[1]。然而,EBI發生、發展的機制目前尚不清楚,研究結果提示,細胞凋亡機制可能參與EBI的復雜分子病理形成過程。本研究結果證實,蛛網膜下腔出血后早期腦皮層血管內皮發生明顯損傷改變,與細胞凋亡信號傳導密切相關的JNK抑制劑SP600125可明顯減輕血腦屏障形態破壞程度,并抑制腦皮層微血管緊密連接相關蛋白Claudin-5和ZO-1表達水平的降低,提示細胞凋亡機制可能參與了EBI復雜分子病理形成過程。
3.2 SAH后早期血腦屏障改變
研究顯示,SAH后早期腦損傷病理生理表現為顱內壓升高,腦灌注壓和腦血流量下降,并引起急性缺血性改變,最主要病理表現為血管源性腦水腫,其機制尚不清楚。腦損傷繼發血管源性腦水腫等改變的分子病理學基礎是血腦屏障的破壞,最關鍵的核心環節是緊密連接相關蛋白結構、功能改變。Claudin-5、ZO-1是分布于腦微血管內皮間最重要的緊密連接相關蛋白,它們的結構、功能異常是血腦屏障破壞的最關鍵環節[4,5]。本研究結果進一步證實,在正常狀態下,腦微血管內皮細胞間連接緊密無開放;蛛網膜下腔出血后,血腦屏障明顯開放,Western blot結果顯示,與sham組相比,SAH大鼠蛛網膜下腔出血后24 h腦皮層緊密連接相關蛋白Claudin-5、ZO-1的表達水平明顯降低(P<0.05),血腦屏障受到破壞。JNK抑制劑SP600125可明顯改善腦皮層微血管及神經元形態學損傷程度,并抑制Claudin-5、ZO-1的表達降低,即抑制血腦屏障開放程度,與透射電鏡觀察到的超微結構的變化相一致。提示大鼠蛛網膜下腔出血后早期血腦屏障結構蛋白發生破壞性改變是早期腦損傷關鍵性機制之一;SP600125可能通過抑制細胞凋亡通路,保護血腦屏障,發揮SAH早期腦損傷的神經保護作用。
本研究結果表明SAH后早期存在以血腦屏障結構破壞為主要特征的腦損傷,細胞凋亡可能參與了SAH后早期腦損傷分子病理學形成過程。
[1]Tseng MY,Czosnyka M,Richards H,et al.Effects of acute treatment with pravastatin on cerebral vasospasm,autoregulation,and delayed ischemic deficitsafter aneurysmal subarachnoid hemorrhage:a phase IIrandomized placebo-controlled trial[J].Stroke,2005,36(8):1627-1632.
[2]Sehba FA,Bederson JB.Mechanismsof acutebrain injury after subarachnoid hemorrhage[J].Neurol Res,2006,28(4):381-398.
[3]RepiciM,Borsello T.JNK pathway as therapeutic target to prevent degenerationinthecentralnervoussystem [J].AdvExp Med Biol,2006,588(1):145-155.
[4]Wolburg H,Lippoldt A.Tight junctions of the blood-brain barrier:development,composition and regulation[J].Vascul Pharmacol,2002,38(6):323-337.
[5]Anderson JM.Molecular structure of tight junctions and their role in epithelial transport[J].News Physiol Sci,2001:16(6):126-130.
[6]Schwartz AY,Masago A,Sehba FA,et al.Experimental models of subarachnoid hemorrhage in the rat:A refinement of the endovascular filament model[J].JNeurosci Methods,2000,96(2):161-167.
[7]Bederson JB,Pitts LH,Tsuji M,et al.Rat middle cerebral artery occlusion:evaluation of the model and development of a neurologic examination[J].Stroke,1986,17(3):472-476.
[8]Kusaka G,Ishikawa M,Nanda A,et al.Signaling pathways for early braininjuryaftersubarachnoidhemorrhage[J].JCerebBloodFlowMetab,2004,24(8):916-925.
[9]Chen D,Nishizawa S,Yokota N,et al.High-dose methylprednisolone prevents vasospasm after subarachnoid hemorrage through inhibition of proteinkinase Cactivation[J].Neurological Res,2002,24(3):215-222.
[10]Chen D,Chen JJ,Yin Q,et al.Role of ERK1/2 and vascular cell proliferation in cerebral vasospasm after experimental subarachnoid hemorrhage[J].Acta Neurochir,2009,151(9),1127-1134.
