孟河醫派大黃煨制工藝優化及炮制后化學成分含量變化研究
2025-01-15 00:00:00薛江林劉雨欣鐘佩劉產明陸兔林李林顏曉靜朱月琴華豐黃瑋
中國藥房 2025年1期
關鍵詞煨大黃;大黃;層次分析;熵權法;正交實驗;成分變化規律;孟河醫派
大黃為蓼科植物掌葉大黃RheumpalmatumL.、唐古特大黃Rheum.tanguticumMaxim.exBalf.或藥用大黃RheumofficinaleBaill.的干燥根及根莖,具有瀉下攻積、清熱瀉火、涼血解毒、逐瘀通經、利濕退黃的功效[1]。生大黃苦寒之性甚強,瀉下作用峻烈,久服易引起腹痛、惡心等胃腸道反應[2],臨床常以炮制品入藥。煨大黃是孟河醫派的特色飲片,大黃經煨制后,瀉下作用減弱,且活血化瘀、消腫止痛作用增強,可治療各類傷癥[3―5],而有緩性增效的作用。
傳統大黃的煨制方法多以濕紙裹后埋入熱火灰中進行加熱,由于火候溫度不可控,導致煨大黃的質量控制較為困難。現代煨大黃研究常以烘箱作為加熱源,采用恒溫方式進行加熱[6],但烘箱中水分蒸發速率過快,與傳統煨制方法存在一定差異。此外,歷版《中國藥典》以及各地方炮制規范均未收錄煨大黃,因此煨大黃的相關炮制方法以及工藝參數尚不明確。基于此,本研究以外觀性狀評分以及5-羥甲基糠醛(5-hydroxymethylfurfural,5-HMF)、沒食子酸、番瀉苷A+番瀉苷B、游離蒽醌、結合蒽醌的含量為評價指標,采用主觀評價的層次分析(analytichierarchyprocess,AHP)法和客觀評價的熵權法結合正交實驗[7],優選大黃最優煨制工藝,同時采用高效液相色譜(HPLC)法對大黃煨制前后的5種蒽醌類成分(蘆薈大黃素、大黃酸、大黃素、大黃酚和大黃素甲醚)、5種蒽醌苷類成分(蘆薈大黃素苷、大黃酸苷、大黃素苷、大黃酚苷和大黃素甲醚苷)、2種番瀉苷類成分(番瀉苷A和番瀉苷B)以及沒食子酸和5-HMF的含量進行對比分析,以期為大黃煨制工藝研究及煨制后化學成分變化規律的闡明提供參考依據。
1 材料
1.1 主要儀器
本研究所用主要儀器有Aglient1260Ⅱ型高效液相色譜儀(美國Aglient公司),XSR105DU型十萬分之一分析天平、ME204型萬分之一分析天平(瑞士Mettler-Toledo公司),IQ7003型Milli-Q純水儀(美國Millipore公司),R201C型旋轉蒸發儀(鞏義市英峪高科儀器廠),DZG-6090型減壓干燥儀(上海培因實驗儀器有限公司)。
1.2 主要藥品與試劑
大黃飲片(批號231024,產地甘肅)購自蘇州市天靈中藥飲片有限公司,經南京中醫藥大學常州附屬醫院藥學部主任中藥師劉產明鑒定為蓼科植物掌葉大黃R.palmatumL.的干燥根和根莖。對照品沒食子酸、5-HMF、番瀉苷A、番瀉苷B、蘆薈大黃素、大黃酸、大黃素、大黃酚、大黃素甲醚(批號分別為B20851、B21832、B20380、B20381、B20772、B20245、B20240、B20238、B20242,純度均大于97%)均購自上海源葉生物科技有限公司;甲醇、碳酸氫鈉、氯仿、磷酸均為分析純;甲醇、四氫呋喃為色譜純;水為純化水。草木灰采自農家土灶,粗草紙購自常州市天寧區農貿市場。
2 方法與結果
2.1 大黃煨制方法
取大黃飲片80g,用濕紙包裹后,放置一定時間,再埋入熱火灰中,然后一并置于烤箱中,先以大火(250~300℃)煨制一定時間后轉文火(120~160℃)煨制,炮制一定時間后取出打開,放涼,即得煨大黃樣品。
2.2 沒食子酸和5-HMF的含量測定方法建立
2.2.1 混合對照品溶液的制備
精密稱取沒食子酸對照品14.30mg、5-HMF對照品14.60mg,加50%甲醇定容于不同10mL容量瓶中,配制成含沒食子酸1.43mg/mL、5-HMF1.46mg/mL的對照品儲備液;分別精密吸取沒食子酸、5-HMF對照品儲備液3.20、0.28mL,混合,加50%甲醇制成沒食子酸和5-HMF質量濃度分別為457.60、40.88μg/mL的混合對照品溶液。
2.2.2 供試品溶液的制備
取生大黃(或煨大黃)粉末0.3g(過四號篩),精密稱定,置具塞錐形瓶中,精密加入50%甲醇25mL,密塞,稱定質量;超聲(功率500W,頻率40kHz,下同)提取30min,放冷,再稱定質量;以50%甲醇補足減失質量,搖勻,濾過,取續濾液過0.22μm微孔濾膜,即得。
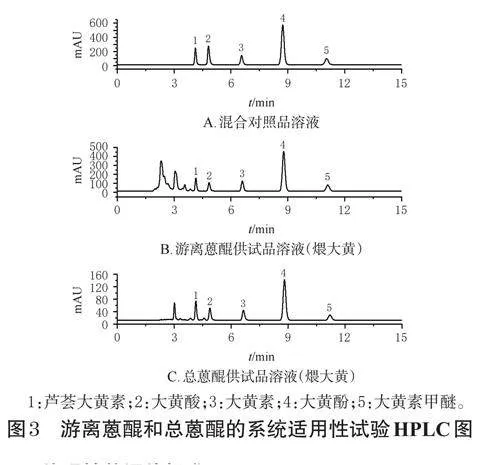
2.2.3 色譜條件、系統適用性試驗與方法學考察
本實驗色譜條件參考文獻[8]設置,以AgilentZorbaxODSC18(250mm×4.6mm,5μm)為色譜柱,以甲醇-0.1%磷酸溶液(V/V,8∶92)為流動相進行等度洗脫,流速為0.8mL/min;柱溫為25℃,檢測波長為273nm,進樣量為10μL,洗脫時間為18min。分別吸取空白對照溶液(50%甲醇)和“2.2.1”“2.2.2”項下混合對照品溶液、煨大黃供試品溶液適量進樣測定,記錄色譜圖。結果顯示,各成分分離度均大于1.5,理論板數按沒食子酸峰計均不低于10000,空白對照溶液對測定無干擾,具體見圖1(空白對照溶液圖略)。參考2020年版《中國藥典》(三部)9101分析方法驗證指導原則[9]進行方法學考察。結果顯示,沒食子酸和5-HMF的回歸方程分別為Y=36.025X-120.490、Y=72.793X-2.000(r≥0.9992),線性范圍分別為3.575~457.600、0.320~40.880μg/mL;精密度試驗、重復性試驗、穩定性試驗(24h)的RSD均小于2.00%(n=6);平均加樣回收率分別為103.3%、98.75%(RSD分別為0.44%、0.36%,n=6)。以上均符合指導原則相關要求。
2.3 番瀉苷A和番瀉苷B的含量測定方法建立
2.3.1 混合對照品溶液制備
精密稱取番瀉苷A對照品7.06mg和番瀉苷B對照品8.59mg,加0.1%碳酸氫鈉溶液定容于不同10mL容量瓶中,配制成含番瀉苷A0.706mg/mL、番瀉苷B0.859mg/mL的對照品儲備液;分別精密吸取番瀉苷A、番瀉苷B對照品儲備液1.40、0.60mL,混合,加0.1%碳酸氫鈉溶液制成番瀉苷A、番瀉苷B質量濃度分別為98.84、51.54μg/mL的混合對照品溶液。
2.3.2 供試品溶液制備
取生大黃(或煨大黃)粉末約0.5g(過四號篩),精密稱定,置于具塞錐形瓶中,加0.1%碳酸氫鈉溶液25mL,稱定質量;超聲提取30min,取出放冷后補足減失質量,搖勻,濾過,取續濾液,過0.22μm微孔濾膜,即得。
2.3.3 色譜條件、系統適用性試驗與方法學考察
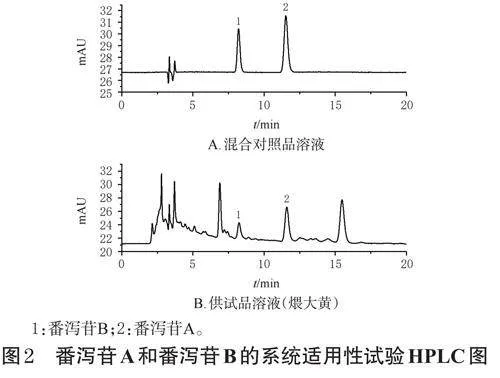
本實驗色譜條件參考文獻[10]設置。以AgilentZorbaxODSC18(250mm×4.6mm,5μm)為色譜柱,以四氫呋喃-水-乙酸(V/V/V,2∶80∶1.5)為流動相進行等度洗脫,檢測波長為380nm,流速為0.8mL/min,柱溫為30℃,進樣量為10μL,洗脫時間為20min。分別吸取空白對照溶液(0.1%碳酸氫鈉溶液)和“2.3.1”“2.3.2”項下混合對照品溶液和供試品溶液(煨大黃)適量進樣測定,記錄色譜圖。結果顯示,各成分分離度均大于1.5,理論板數按番瀉苷B峰計均不低于7000,空白對照溶液對測定無干擾,具體見圖2(空白對照溶液圖略)。參考2020年版《中國藥典》(三部)9101分析方法驗證指導原則[9]進行方法學考察。結果顯示,番瀉苷A和番瀉苷B的回歸方程分別為Y=7.6407X-1.9066、Y=8.5195X-1.3115(r≥0.9999),線性范圍分別為3.090~98.840、1.610~51.540μg/mL;精密度試驗、重復性試驗、穩定性試驗(24h)的RSD均小于2.00%(n=6);平均加樣回收率分別為99.11%、97.12%(RSD分別為1.79%、1.32%,n=6)。以上均符合指導原則相關要求。
2.4 游離蒽醌和結合蒽醌的含量測定方法建立
游離蒽醌和總蒽醌的含量均以蘆薈大黃素、大黃酸、大黃素、大黃酚和大黃素甲醚5種成分的總量計,結合蒽醌的含量以蘆薈大黃素苷、大黃酸苷、大黃素苷、大黃酚苷和大黃素甲醚苷5種成分的總量計(但定量分析時,各蒽醌苷類成分含量以對應苷元含量計),其中結合蒽醌含量=總蒽醌含量-游離蒽醌含量。
2.4.1 混合對照品溶液的制備
精密稱取蘆薈大黃素對照品0.40mg、大黃酸對照品0.84mg、大黃素對照品0.52mg、大黃酚對照品1.84mg、大黃素甲醚對照品0.44mg,加甲醇定容至同一10mL容量瓶中,制成含蘆薈大黃素0.040mg/mL、大黃酸0.084mg/mL、大黃素0.052mg/mL、大黃酚0.184mg/mL、大黃素甲醚0.044mg/mL的混合對照品溶液。
2.4.2 供試品溶液制備
(1)游離蒽醌供試品溶液的制備:取生大黃(或煨大黃)粉末約0.5g(過四號篩),精密稱定,置于具塞錐形瓶中,精密加入甲醇25mL,稱定質量;加熱回流1h,放冷,再次稱定質量,用甲醇補足減失的質量,搖勻,濾過,取續濾液,過0.22μm微孔濾膜,即得。(2)總蒽醌供試品溶液的制備:取生大黃(或煨大黃)粉末約0.15g(過四號篩),精密稱定,置于具塞錐形瓶中,精密加入甲醇25mL,稱定質量;加熱回流1h,放冷,再次稱定質量,用甲醇補足減失的質量,搖勻,濾過;精密量取續濾液5mL,置于燒瓶中,揮去溶劑,加8%鹽酸溶液10mL,超聲處理2min;再加三氯甲烷10mL,加熱回流1h,放冷,置于分液漏斗中,再用少量三氯甲烷洗滌容器,洗滌液并入分液漏斗中,取三氯甲烷層;再用三氯甲烷提取3次,每次10mL,合并三氯甲烷液,減壓回收溶劑至干;殘渣加甲醇使溶解,轉移至10mL容量瓶中,加甲醇至刻度,搖勻,濾過,取續濾液,過0.22μm微孔濾膜,即得。
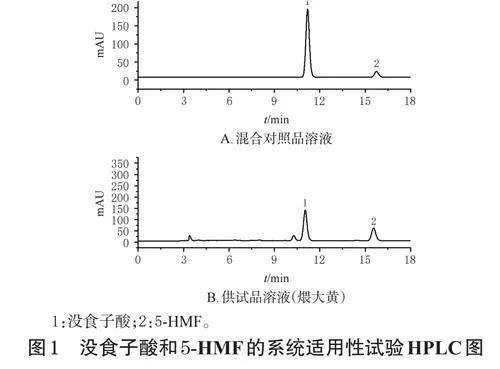
2.4.3 色譜條件、系統適用性試驗與方法學考察
本實驗色譜條件參考文獻[11]設置。以AgilentZorbaxODSC18(250mm×4.6mm,5μm)為色譜柱,以甲醇-0.1%磷酸溶液(V/V,85∶15)為流動相進行等度洗脫,檢測波長為254nm,流速為1.0mL/min,柱溫為30℃,進樣量為10μL,洗脫時間為15min。分別吸取空白對照溶液(甲醇)和“2.4.1”“2.4.2”項下混合對照品溶液、煨大黃游離蒽醌和總蒽醌供試品溶液適量進樣測定,記錄色譜圖。結果顯示,各成分分離度均大于1.5,理論板數按大黃酸峰計均不低于9000,空白對照溶液對測定無干擾,具體見圖3(空白對照溶液圖略)。參考2020年版《中國藥典》(三部)9101分析方法驗證指導原則[9]進行方法學考察,結果顯示,蘆薈大黃素、大黃酸、大黃素、大黃酚和大黃素甲醚的回歸方程分別為Y=44.288X+4.203、Y=28.356X+1.587、Y=27.553X+1.356、Y=39.703X+20.806、Y=32.660X+1.431(r≥0.9999),線性范圍分別為0.020~0.480、0.042~1.008、0.026~0.624、0.092~2.208、0.022~0.528μg;精密度試驗、游離蒽醌的重復性試驗和穩定性試驗(24h)、總蒽醌的重復性試驗和穩定性試驗(24h)的RSD均小于3.00%(n=6);游離蒽醌的平均加樣回收率分別為97.35%、101.94%、100.89%、98.40%、99.50%(RSD分別為0.69%、3.05%、1.85%、0.66%、0.87%,n=6);總蒽醌的平均加樣回收率分別為98.01%、100.97%、97.16%、98.59%、97.38%(RSD分別為1.02%、3.32%、2.28%、0.65%、1.27%,n=6)。以上均符合指導原則相關要求。
2.5 外觀性狀評價標準
筆者根據古籍中“大黃濕紙裹煨勿令焦”“大黃濕紙包,煨令香熟”“濕紙裹煨至紙焦”等相關描述,并參考相關文獻的評分方法[6,11],對煨大黃的紙張性狀、飲片氣味、顏色以及質地進行評分,具體標準為:當飲片濃香、較硬、呈深棕黃色且紙焦時,記為3分;當飲片清香、較軟、呈黃褐色且紙干時,記為2分;當飲片焦香、輕脆、呈焦褐色且紙濕時,記為1分。
2.6 AHP-熵權法計算煨大黃的綜合評分
2.6.1 AHP確定權重rj
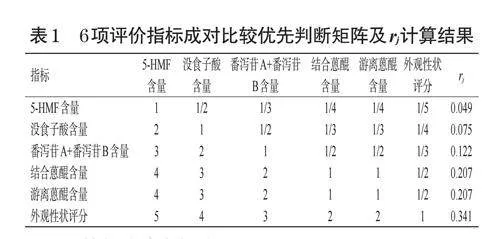
煨大黃作為古代應用普遍但現代已經失傳的炮制品種,外觀性狀特征對其質量評價具有重要作用,因此在AHP中設置最高權重;游離蒽醌是2020年版《中國藥典》中大黃質量評價的重要指標,也是大黃發揮活血化瘀作用的主要藥效成分[12];結合蒽醌和番瀉苷類是大黃及其炮制品發揮瀉下作用的主要藥效成分[13―14];沒食子酸是大黃發揮收斂止血、抗炎作用的主要藥效成分,且大黃煨制后該成分含量變化較大[15];5-HMF具有多種藥理活性但有一定毒副作用[16],故在AHP中設置最低權重。最終確認各指標重要程度依次為外觀形狀評分>游離蒽醌含量=結合蒽醌含量>番瀉苷A+番瀉苷B含量>沒食子酸含量>5-HMF含量,構建各指標的優先判斷矩陣,并根據方根法計算各指標的rj,具體見表1。進一步對矩陣進行一致性檢驗發現,一致性參數λmax=6.072,一致性指標=0.014,一致性比率=0.011<0.1,表明矩陣合理有效,權重系數可靠。
2.6.2 熵權法確定權重wj
將測得數據代入相應公式計算,其中,正向指標代入Yij=(Xij-Xmin)/(Xmax-Xmin),負向指標代入Yij=(Xmax-Xij)/(Xmax-Xmin)中,式中,Yij為j指標下第i個數值的標準化值,Xij為第i次實驗時第j個指標的測定值,Xmin為該組測定值中的最小值,Xmax為該組測定值中的最大值。將標準化數據矩陣(Yij)轉化為概率矩陣(Pij),Pij=Yij/Σi=1nYij;再求各指標信息熵(Hj),Hj=-kΣi=1nPijlnPij,k=1/lnn(n為樣本量);然后采用熵權法計算各指標權重wj,wj=(1-Hj)/Σj=1m(1-H)j。
2.6.3 綜合評分的計算方法
用AHP法得到的rj、熵權法得到的wj,計算綜合權重(Fj),Fj=rjwj/Σj=1mrjwj。結果顯示,外觀性狀評分和5-HMF、沒食子酸、番瀉苷A+番瀉苷B、游離蒽醌、結合蒽醌含量的Fj分別為0.235、0.115、0.068、0.182、0.156、0.244,然后計算綜合評分(M),M=0.235×100×外觀性狀評分/外觀性狀評分最大值+0.115×100×5-HMF含量/5-HMF含量最大值+0.068×100×沒食子酸含量/沒食子酸含量最大值+0.182×100×(番瀉苷A+番瀉苷B)含量最小值/(番瀉苷A+番瀉苷B)含量+0.156×100×游離蒽醌含量/游離蒽醌含量最大值+0.244×100×結合蒽醌含量最小值/結合蒽醌含量。
2.7 大黃煨制工藝的優選
結合本研究前期預實驗,選取文火溫度、煨制時間、紙張層數、紙裹時間為考察因素,以外觀性狀評分以及沒食子酸、5-HMF、番瀉苷A+番瀉苷B、結合蒽醌、游離蒽醌含量為評價指標,采用AHP-熵權法計算綜合評分M,再根據正交實驗優選大黃煨制工藝。
2.7.1 正交實驗設計與結果
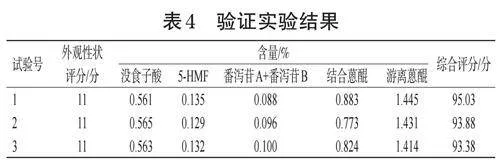
根據預實驗中不同煨大黃飲片的外觀性狀篩選出部分適宜條件:固定大火溫度加熱時間為20min,文火溫度(A)取120、140、160℃3個水平,煨制時間(B)取1.5、2、2.5h3個水平,紙張層數(C)取1、2、3層3個水平,紙裹時間(D)取0、0.5、1h3個水平,進行4因素3水平的L(934)正交實驗。具體方案與結果見表2,方差分析結果見表3。
由表2、表3可知,各因素影響大小順序為B>C>A>D,且因素A、B、C對煨大黃質量均有顯著影響,因此最終確定最優的工藝為A2B3C1D2,即每80g大黃用1層濕紙包裹0.5h,大火煨制20min后轉文火140℃煨制,總煨制時間2.5h。
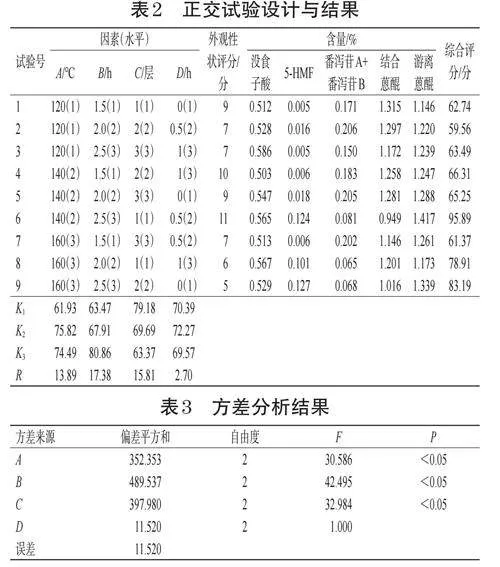
2.7.2 驗證實驗
按“2.7.1”項下最優工藝制備煨大黃樣品,進行3次驗證實驗。結果顯示,3次驗證實驗的綜合評分均值為94.10,RSD小于1.0%,表明優化工藝穩定、可行。驗證實驗結果見表4。
2.8 大黃煨制后化學成分變化研究
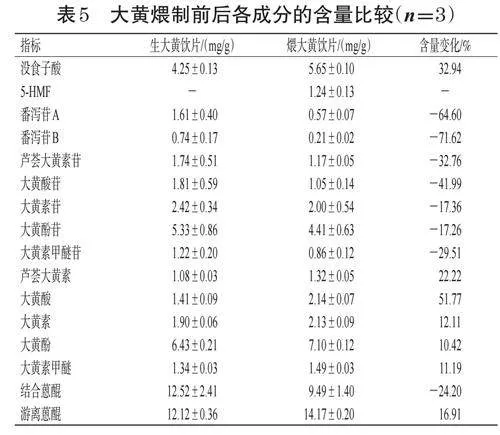
取生大黃飲片及“2.7.1”項下按最優工藝制備煨大黃飲片適量,按“2.2”~“2.4”項下方法測定其中5種蒽醌類成分、5種蒽醌苷類成分、2種番瀉苷類成分以及沒食子酸和5-HMF的含量(平行測定3次),并分析含量變化情況[含量變化=(炮制后成分含量-炮制前成分含量)/炮制前成分含量×100%]。結果顯示,大黃煨制后5種蒽醌苷類成分和2種番瀉苷類成分的含量均大幅降低,而5種游離蒽醌類成分和沒食子酸含量均不同程度升高,且新生成了5-HMF,表明煨制過程會導致大黃化學成分發生轉化。結果見表5。
3 討論
《通俗文》記載的“熱灰謂之煻煨”,是將藥物包裹后置于炭火或柴火的余燼中緩慢加熱至一定程度的炮制方法。孟河醫派古籍中也以“熱火灰煨”或“煻火煨”描述煨法。這種煨制方法工藝繁瑣,條件不易控制,且有安全隱患,不適于現代生產。本課題組前期比較了目前主流的濕紙裹煨法、濕紙裹后烘箱直接加熱法以及熱沙煨制法[6,17],發現這些方法煨制的大黃與古籍中煨大黃的性狀記載均有較大差異,因此本研究選擇保溫保濕性能較好的熱火灰,用烤箱代替火源,并模擬古法炮制真實的溫度變化,采取先大火加熱后文火加熱的方式進行煨制,并在此基礎上對大黃的煨制工藝進行優化,結果顯示,大黃的最優煨制工藝為:每80g大黃用1層濕紙包裹0.5h,大火煨制20min后轉文火140℃煨制,總煨制時間2.5h。
大黃煨制后5種蒽醌類成分、5種蒽醌苷類成分、2種番瀉苷類成分以及沒食子酸和5-HMF的含量均發生明顯變化,其中蒽醌苷類和番瀉苷類成分含量大幅降低,而游離蒽醌含量升高,這可能是由于大黃煨制過程中,番瀉苷類成分結構被破壞,蒽醌苷類成分受熱易分解為相應的苷元;沒食子酸含量的升高可能是大黃中縮合鞣質與可水解鞣質的化學鍵在高溫下斷裂形成了單體成分沒食子酸[18]。5-HMF是大黃中糖類成分發生焦糖化反應的產物,一定程度上可以反映炮制程度和產品質量,且具有抗氧化、改善血液流變學等藥理活性[8,16]。研究表明,蒽醌類成分中的糖苷鍵可以保護苷元不被小腸水解吸收以順利進入結腸,刺激腸道而致瀉[19];番瀉苷A可通過調控腸道菌群和水通道蛋白表達發揮瀉下作用[20];大黃素、蘆薈大黃素、大黃素甲醚等可作用于內皮型一氧化氮合酶等關鍵靶點,發揮活血化瘀作用[12];沒食子酸對角叉菜膠誘導的急性炎癥大鼠具有良好的抗炎消腫止痛作用[21]。由此表明,大黃煨制過程中,蒽醌苷類和番瀉苷類成分的分解和破壞可能是煨大黃苦寒瀉下作用減輕的主要原因,而沒食子酸和游離蒽醌含量的升高可能是煨大黃發揮活血化瘀作用的物質基礎,這也與煨大黃在孟河醫派中的臨床應用一致。
綜上所述,本研究成功優化了大黃的煨制工藝,并分析了大黃煨制后的化學成分含量變化,可為大黃煨制工藝的深入研究及煨制過程中化學成分變化規律的闡明奠定基礎。
