序列比對算法BWA的優化
2024-12-30 00:00:00胡爽陳長波
計算機應用研究 2024年12期








摘 要:序列比對是基因組數據分析的關鍵一環,提高其比對效率對推動測序技術在醫學、古生物學等領域的應用具有重要意義。針對BWA算法兩大步驟之一的SAMSE存在冗余讀取索引導致效率欠佳的問題,提出了新的算法BWA*。其通過運用流程優化消除了SAMSE中參考序列及其索引的冗余讀取,在此基礎上疊加運用關鍵參數值調整和多線程優化等技術,進一步提高了計算效率。公開數據庫中的真實序列測試表明,BWA*的SAMSE步驟的性能是BWA中SAMSE性能的7.11~8.61倍,平均為7.84倍,BWA*的整體性能是BWA的1.25~1.70倍,平均1.47倍。針對實際應用中的古代DNA序列比對,實驗表明和另一常用工具BWA-MEM相比,優化后的BWA*在繼承原有BWA高精度特性的同時,實現了對BWA-MEM速度的超越。
關鍵詞:序列比對;羅伯斯-惠勒變換;第二代測序;BWA;古代DNA
中圖分類號:R857.3"" 文獻標志碼:A
文章編號:1001-3695(2024)12-034-3777-09
doi: 10.19734/j.issn.1001-3695.2024.05.0157
Optimization of sequence alignment algorithm BWA
Hu Shuang1, 2, 3, Chen Changbo1, 3
(1.Chongqing Key Laboratory of Secure Computing for Biology,Chongqing Institute of Green amp; Intelligent Technology, Chinese Academy of Sciences, Chongqing 400714, China; 2. School of Computer Science amp; Technology, Chongqing University of Posts amp; Telecommunications, Chongqing 400065, China; 3. Chongqing School, University of Chinese Academy of Sciences, Chongqing 400714, China)
Abstract:Sequence alignment is a critical component of genomic data analysis, and improving its efficiency is vital for advancing sequencing technology applications in medicine, paleobiology, and other fields. This paper addressed the inefficiency in the SAMSE step of the BWA algorithm, caused by redundant index reads, by proposing a new algorithm called BWA*. Through process optimization, BWA* eliminated redundant reads of reference sequences and their indices in the SAMSE step. Additionally, the algorithm incorporated key parameter adjustments and multithreading optimizations to further enhance computational efficiency. Tests on real sequences from public databases show that the performance of BWA* in the SAMSE step is 7.11 to 8.61 times that of BWA, with an average of 7.84 times. Overall, BWA* achieves 1.25 to 1.70 times the perfor-mance of BWA, with an average of 1.47 times. In practical applications of ancient DNA sequence alignment, experiments demonstrate that the optimized BWA* surpasses the speed of BWA-MEM while maintaining the high accuracy characteristic of the original BWA.
Key words:sequence alignment; Burrows-Wheeler transform(BWT); next generation sequencing; Burrows-Wheeler aligner; ancient DNA
0 引言
自人類基因組計劃成功實施以來,基因組測序技術得到了迅速發展。NGS (next-generation sequencing) 技術[1] (又稱高通量測序技術)的發展使人類基因組測序的成本從2007年的100萬美元下降到2022年的100美元[2],花費的時間也從Sanger測序時代的幾年降低到了5個小時[3]。測序成本的降低和數據生成速度的提高使得基因組序列數據呈爆炸式增長,并進一步推動了生命科學的研究。NGS產生的海量測序數據需要進行序列比對[4],即將讀取到的序列比對到參考序列上,以確定讀取序列位置,并推斷序列間的差異[5]。通過序列比對,可以幫助揭示不同生物體或樣本之間的遺傳信息相似性和差異性,從而幫助研究人員進行生物學、進化學和醫學等方面的研究。序列比對通常是NGS數據分析中花費時間最久的一個步驟[6, 7],因此加速序列比對的速度具有重要的意義。序列比對算法根據其索引方式主要可以分為基于哈希表的和基于BWT的方法。哈希表的方法[8]基于哈希參考基因組的索引,以獲得更精確的比對結果。該設計為reads找到其所有的種子,定位到種子在reference序列中出現的位置,然后在這些位置執行Smith-Waterman比對[9]。
BWT[10]最初被用作文本壓縮算法,主要思想是把一個字符串中的相同字符聚集在一起。結合基于FM-index[11]的搜索技術,與基于哈希表的算法相比,基于BWT的算法內存占用效率更高,而且不會影響速度[12]。該算法被基因組界廣泛采用,用于開發高效的比對技術。
BWA(Burrow-Wheeler aligner)[13]是基于BWT的一組用于基因組序列比對算法的統稱,包含了BWA-BACKTRACK、BWA-SW和BWA-MEM[14]三個算法。因為BWA-BACKTRACK是BWA工具包中最先被開發出的算法,一開始只有這一個算法,所以文獻中一般也將BWA-BACKTRACK直接稱為BWA。本文遵循慣例,后面皆用BWA作為BWA-BACKTRACK的簡稱。BWA在2009年被開發出來后,就開始被生物基因等相關領域廣泛使用,而它至今依然被廣泛使用,主要原因是它可以適應多樣化的數據。BWA被設計用于處理比較短(100 bp以下)的測序數據。特別地,在古生物學研究中,由于古DNA樣本通常是短片段,BWA可以有效地比對古DNA序列,幫助研究人員揭示古代生物的遺傳信息、進化歷史和種系關系[15~17]。
雖然現代測序技術不斷發展,但因為三代測序技術錯誤率高和成本高的問題[18],產生短序列的二代測序技術仍普遍適用于常規基因組測序,所以包括BWA、BWA-MEM等在內的基于BWT的序列比對算法仍然適用于很多的應用。BWA的說明文檔中表明,相對于BWA,BWA-MEM更常用于70 bp到1 mbp,而BWA一般用于100 bp以下的序列[19]。
本文的主要貢獻是針對BWA算法兩大主要步驟之一的 SAMSE效率欠佳的問題,提出了五步遞進優化,在讀長為35 bp~100 bp,測序片段數(number of reads)為千萬的不同測序序列上驗證了其效率優于原始的BWA,具體貢獻如下:
通過優化算法流程,將SAMSE中重復讀取參考序列索引和參考序列的步驟分別優化為各自僅需讀取一次。實驗表明,通過去除參考序列索引冗余讀取,SAMSE性能提升為原始SAMSE的1.59~1.83倍,平均為1.68倍,通過去除參考序列冗余讀取,將SAMSE性能進一步提高為原始SAMSE的1.71~2.15倍,平均為1.87倍。
通過實驗分析,為去除讀取冗余后的SAMSE的后綴數組索引參數SA_INTERVAL提出了更合理的默認值。實驗表明,使用新SA_INTERVAL值的SAMSE性能進一步提升為原始SAMSE的2.70~3.38倍,平均為3.12倍。
通過將上述優化后的SAMSE中的關鍵計算步驟改造為多線程實現,進一步提高了在多核CPU處理器上的計算效率。實驗表明,多線程優化(4線程)后的SAMSE性能進一步提高為原始SAMSE的6.67~8.15倍,平均為7.34倍。
通過實驗分析,為多線程優化后的SAMSE的堿基順序出現頻率參數OCC_INTERVAL提出了新的默認值。實驗表明,運用新的OCC_INTERVAL值的SAMSE的性能進一步提高為原始SAMSE的7.11~8.61倍,平均為7.84倍。
通過將原始BWA中的SAMSE替換為五步優化后的SAMSE,并通過實驗分析進一步優化OCC_INTERVAL的取值,得到改進后的算法BWA*。實驗表明,BWA*性能是原始BWA的1.25~1.70倍,平均1.47倍。在長度為35 bp,reads大于兩百萬條上的測試表明,性能相較于 BWA,BWA*對 BWA-MEM 的性能優勢,從1.31~1.46倍,平均為1.40倍,提高到了1.91~2.67倍,平均為2.34倍。
1 相關工作和背景知識
1.1 相關工作
序列比對算法的研究進程按照基因測序技術來劃分主要分為以下幾個階段:
a)第一代測序技術背景下的序列比對算法。第一代測序技術即Sanger測序,這種測序方式是分段測序的,測序長度可達900 bp、測序精度高可達99.9%,但是因為成本太過高昂而無法大規模應用[20]。在這一背景下,當時最具代表性的算法是BLAST和FASTA算法,這兩種算法引入了一些啟發式算法和局部近似比對的方法,并通過預處理和構建索引來加速比對過程,讓序列比對有了處理大規模數據的可能。
b)第二代測序技術背景下的序列比對算法。第二代測序技術也稱高通量測序技術即Illumina等測序技術,二代測序技術實現了并行測序,它能在同一時間對多個DNA片段進行測序,因此二代測序可以產生大量的短讀序列,通常為幾十到幾百個堿基對,并且測序成本大大降低,推動了基因組學研究的廣泛運用。處理二代測序產生的短讀序列所用的序列比對算法主要是基于BWT構建索引的Bowtie、Bowtie2、BWA及BWA-MEM等。BWT算法的核心思想是將一個字符串轉換為另一種形式,讓相同的字符聚集在一起,從而提高壓縮和搜索效率。在短序列的基因組分析過程中,序列比對是計算成本最高的步驟[4]。
c)第三代測序技術背景下的序列比對算法。第三代測序技術,如PacBio[21]和Oxford Nanopore等測序技術。主要測的是比較長的序列段,長度通常為幾千甚至幾萬個堿基對。為了比對這種長序列,誕生了很多長序列比對算法,如利用哈希函數的Minimap[22]和使用圖數據結構進行序列比對的GraphMap算法。但是三代測序技術仍然存在一些問題,如測序的錯誤率在90%左右,雖然某些序列可以達到98%,但是也需要額外花費高昂的計算成本。總的來說,三代測序技術還沒有二代測序那樣穩定而精確[6,16],所以目前比較成熟的二代測序技術仍然是研究者優先選擇的對象。
目前對于Bowtie2和BWA這類針對二代測序序列且基于BWT的主流序列比對算法的改進有很多。Pham-Quoc等人[23]實現了BWA-MEM與FPGA的結合,在性能上獲得了極大的提升;文獻[24]開發的最新版本的BWA-MEM2利用多核處理器加速了BWA-MEM,比BWA-MEM快了2.07倍;Pham等人[25]實現了BWA-MEM與GPU的結合,將BWA-MEM的比對速度提升至了最新的BWA-MEM2的2.5倍;Song等人[26]提出了一種基于強化學習的局部比對方法來獲得最優序列比對,將計算的復雜度大大降低;Roodi等人[27]調整了BWA-MEM中隱含的參數SA_INTERVAL,研究了參數對內存和比對速度的影響,將整個人類基因組SA索引讀入了內存,最終速度提高了19%。但是其并沒有繼續研究OCC_INTERVAL的影響,而且沒有將這個參數推廣到同樣使用BWT算法生成索引的BWA等比對算法上。
1.2 BWT介紹
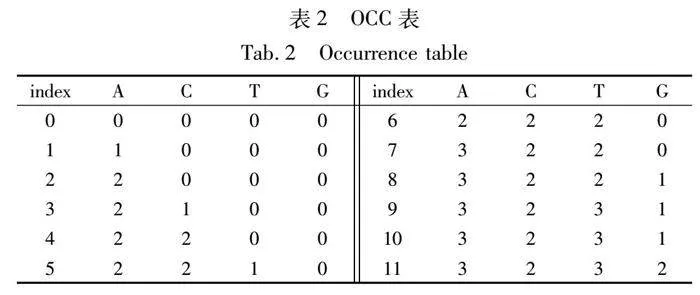
在介紹本文工作之前首先介紹一下BWT和它是如何構建索引的,以BWT和FM-index(full-text index in minute space)[28]等作為索引方式的算法生成的索引:BWT字符串、出現頻率表(occurrence table,OCC)和后綴數組(suffix array,SA)幾個部分。
1.2.1 BWT和SA數組
在BWA的比對過程中首先要對參考序列進行BWT變換,具體步驟如下:
1.2.4 利用C表和OCC表進行反向搜索
利用前面的count表和occurrence表,再加上top和bottom兩個索引指針,就可以實現比對搜索[28]。
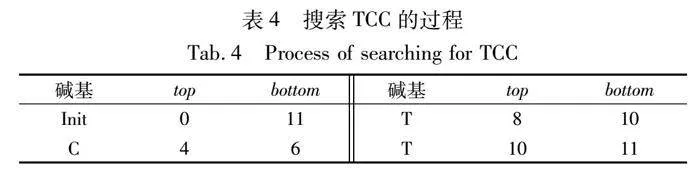
在長度為n的reference序列R中搜索一個長度為m的read序列Q,BWT比對算法從右向左逐個搜索Q的堿基。top初始化為0,bottom初始化為n+1。在從右向左逐個字符搜索序列Q的過程中,根據式(1)(2),通過訪問count表和OCC表來更新top和bottom兩個指針的值:
topnew=count(bp)+occ(topold,bp)(1)
bottomnew=count(bp)+occ(bottomold,bp)(2)
因為是從右向左逐個字符進行搜索的,式中bp代表的是正在搜索的字符。最終得到的top和bottom代表的是Q在BWT矩陣中第top行到第bottom-1行作為前綴,如果top≥bottom則說明沒有比對上;如果top=bottom-1,則說明比對上了一個位置;如果toplt;bottom-1,則說明比對上了多個位置。搜索完成后通過SA表來檢索Q在參考序列R上的實際位置。
表4是在R=TCAGGTTCAA中搜索Q=TTC,top和bottom指針更新的例子,需要注意的是搜索得到的區間是左閉右開的,即[10,11),表明TTC在BWT矩陣的第10行。接下來查看SA表的第10行,得到在參考序列R上的位置為5,即TTC比對上了參考序列R上第5個字符。
而如果搜索的是TC則只需比對到第二行得到[8,10),即TC在BWT矩陣的第8、9兩行。然后查看SA表的第8、9行,得到在參考序列R上的位置為0和6,即TC比對上了參考序列R上第0個和第6個字符上。如果在R上搜索GC,搜索過程如表5所示:比對搜索的結果為[6,6)說明R上并沒有找到GC。
而實際比對的過程并不是這樣的精確比對,BWA允許一定的錯配和空位,這種策略使其在處理序列變異和錯誤方面表現出較高的靈活性和準確性。
2 本文方法
本文的主要工作分為三個方面:a)消除了BWA的SAMSE步驟中由于反復讀取參考序列及索引信息產生數據冗余讀取問題,提高了BWA的比對速度;b)將改進后的BWA的SAMSE部分進行了多線程并行實現,使得BWA整體可并行,進一步提高了比對速度;c)研究了兩個重要參數SA_INTERVAL和OCC_INTERVAL對改進后的BWA性能的影響,重新給出了默認推薦參數值。
2.1 BWA算法流程的優化
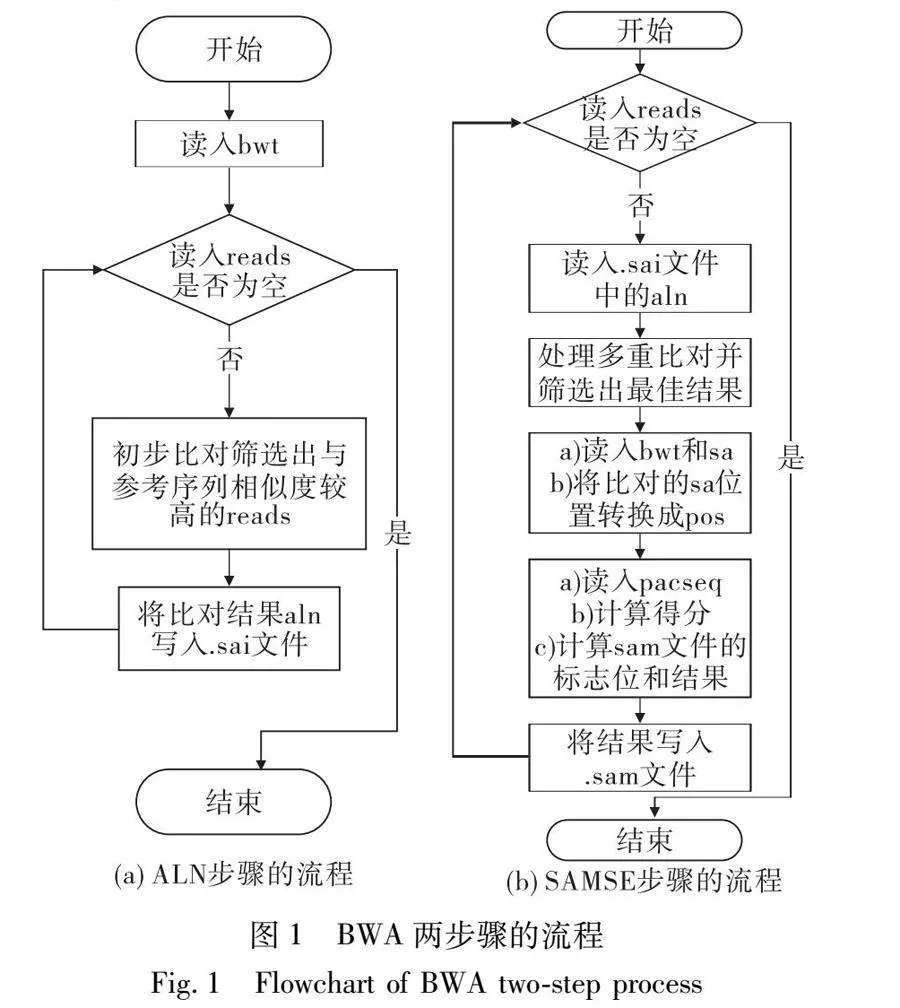
從原理上講,BWA算法主要有構建索引、初步篩選合適的比對位置和比對并計算結果三個步驟。在實現上BWA分成ALN和SAMSE兩步對應初步篩選和計算結果兩步,對算法抽象可以得到如圖1中兩個流程,ALN步驟由初始化各種數據結構、讀入BWT然后將reads按批次讀入比對并寫入.sai文件中直至讀完,這幾步構成。而SAMSE步驟主要分為:初始化、將reads按批循環讀入并提取比對信息,然后計算生成結果并寫入.sam文件中。
通過這個抽象,本文對開銷最大的部分仔細分析可以看實際上它是由ALN部分的比對步驟和SAMSE部分的計算結果步驟兩部分組成。對SAMSE這部分的功能可以拆分成從硬盤中讀取計算所需的信息和計算結果信息兩部分。其中讀取bwt、sa表和讀取pacseq(參考序列),這兩部分每一次循環都會讀入一次,SA是BWT索引的一部分,是用來記錄參考序列子序列位置的信息,而還原sa表也需要bwt,參考序列pacseq則是在KSW2算法中用于比對并計算分數的。而算法并不會對sa表、bwt和pacseq做任何修改,也就是說索引重復讀入sa表和bwt其實是冗余的。其中值得注意的是,循環的次數是由reads數決定的,因為每次循環會讀入固定數量的reads,直至所有的reads都被讀完,所以reads中包含的read越多,這種冗余就越多。因此不需要將它放在循環中多次運行。通過拆分可以得到一個新的流程如圖2所示,可以將讀取sa表、bwt和讀入pacseq的操作提取到循環之前來,只需要讀入一次。
2.2 參數對BWA的影響
SA_INTERVAL和OCC_INTERVAL是BWA中隱含的參數,在生成索引時,索引中的SA和OCC值生成了一部分,每隔SA_INTERVAL行或OCC_INTERVAL行保存一行數據,也就是只保存checkpoint位置的數據。可以說SA_INTERVAL和OCC_INTERVAL決定了生成的索引的大小,同樣在比對的過程中索引是要讀入內存中的,所以這兩個參數也影響著讀數據的時間和占用內存的大小。
而在比對的過程中需要花費時間還原需要用到的SA表和OCC表:在ALN步驟中,在參考序列上查找reads可能比對的位置,需要反復調用OCC表來快速定位,這時就需要計算和還原OCC表中未被保存行的信息;而在SAMSE步驟中需要利用SA表,將在ALN步驟中查找到的SA位置還原成在參考序列上的位置,因此也需要計算和還原SA表中未被保存行的信息。所以說SA_INTERVAL和OCC_INTERVAL也影響著比對的時間,選擇合適的參數對比對效率的影響至關重要。
基于前面介紹的BWT變換的過程,以及各個表和數據結構,接下來介紹這兩個參數具體是如何影響序列比對的。
2.2.1 后綴數組SA的還原
式(3)中的(ψ-1)j代表使用了j次ψ-1,ψ-1如式(4)所示,它的作用是找到當前的字符B(i)在原序列中的前一個字符在SA表中的行數,通過對這個式子迭代j次找到已保存的SA的行數,這一行的SA加上迭代的次數就是要還原位置的SA[11]。
S(k)=S((ψ-1)j(k))+j(3)
ψ-1(i)=count(B(i))+occ(B(i),i)(4)
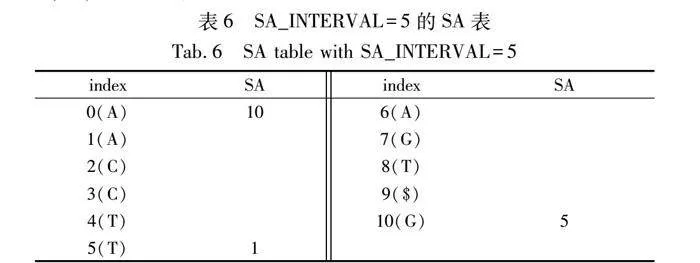
下面對2.2.1節中的示例參考序列 R=TCAGGTTCAA,通過OCC表和C表還原SA表中空缺值:假設SA_INTERVAL = 5,如表6所示,只有0、5和10三行的SA,如果需要還原第2行的SA,根據式(3)(4)進行迭代。
第一次:
ψ-1(2)=count(B(2))+occ(B(2),2)=count(C)+occ(C,2)=4+0=0,S(2)=S(4)+1
第二次:
ψ-1(4)=count(B(4))+occ(B(4),4)=count(T)+occ(T,4)=8+0=8,S(4)=S(8)+1
第三次:
ψ-1(8)=count(B(8))+occ(B(8),4)=count(T)+occ(T,8)=8+2=10,S(4)=S(10)+1
此時S(10) 是已知的,為5,因為迭代了三次,所以S(2) = S(10) + 3 = 8。
2.2.2 OCC表的還原
索引中的OCC信息也保存了部分,每隔OCC_INTERVAL行保存一行,保存的那些行叫做checkpoint[11]。而當比對中需要用到未保存的OCC信息時,則需要還原需要那一行的OCC信息,方法是先找到那一行之前最近的checkpoint,通過B得到此checkpoint到那一行之間各堿基出現了多少次,與checkpoint分別相加或相減就得到了那一行所有的OCC信息。
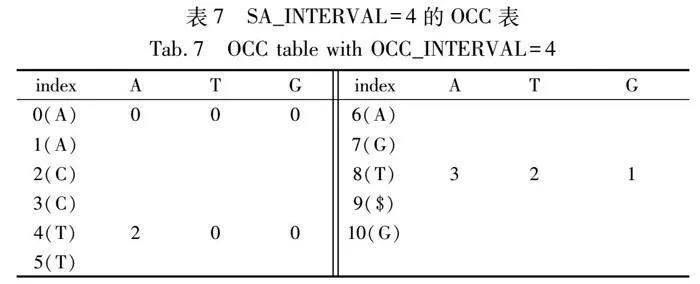
表7是前面R=TCAGGTTCAA的例子,假設OCC_INTERVAL = 4時的OCC表,如果需要occ(T,6),就可以先找到最近的checkpoint如index=4那一行,查看BWT序列發現 [4,6) 行有兩個T,于是就可以得到occ(T,6)=occ(T,4)+2=2。
2.2.3 參數對BWA和BWA-MEM的影響
通過實驗可以發現SA_INTERVAL和OCC_INTERVAL對BWA和BWA-MEM的比對時間和內存占用都有一定影響。因為以FM-index方式生成索引的軟件,如BWA、BWA-MEM等序列比對算法只生成部分索引,而其他索引信息則在比對過程中通過這些部分索引動態生成所需的索引信息。因此生成的索引越多,讀入索引的時間就越多,所需的內存也越多,還原索引的計算就越少;相反生成的索引越少,讀入索引的時間就越少,所需的內存也越少,還原索引的計算就越多。
2.3 多線程實現
原始BWA中ALN步驟是支持多線程的,但是SAMSE步驟并不支持多線程,不能充分利用多核處理器等計算資源。BWA-MT對原始BWA算法中的SAMSE步驟進行了多線程實現,但其并沒有提前去除算法流程上的冗余。本文基于pthread對SAMSE的改進版本samse3進行了多線程優化。算法根據用戶輸入的線程數,將reads分成幾個子集,然后線程并行地處理各部分reads。
算法1 bwa_sai2sam_se_core()
輸入:.sai文件、reads。
輸出:.sam文件。
1 while ((讀入n_seqs個reads) is not 1)
2" for i = 0 to n_seqs-1
3"" //逐條處理每條reads對應的比對
4"" bwa_aln2seq_core();
5"" //篩選符合條件的比對并計算比對的錯配數、gap數和
6"" //比對位置在參考序列上的偏移量
7" end for
8" bwa_cal2seq_core();
9" //將sa位置轉換成pos
10 "bwa_refine_gapped();
11 "http://計算CIGAR、MD,使用KSW2算法計算比對分數
12 "for i = 0 to n_seqs-1
13"" bwa_print_sam1();
14"" //將計算的結果信息寫入到.sam文件中
15" end for
16 end while
如算法1所示,bwa_sai2sam_se_core是一個在SAMSE步驟中計算比對結果信息的核心函數。首先,它讀取n_seqs個reads,并通過bwa_aln2seq_core這個函數根據比對結果的質量篩選出符合條件的比對結果,然后bwa_cal_pac_pos函數將.sai文件中比對位置的SA坐標轉換為序列坐標。bwa_refine_gapped計算了比對的CIGAR字符串,最后將結果轉換為SAM文件格式。重復這些處理步驟,直到處理完所有的輸入讀序列。但是該算法和實現被設計用于單個處理器核心。于是本文對SAMSE步驟進行了多線程實現。如算法2所示,首先將SAMSE步驟中的主要函數bwa_aln2seq_core、bwa_cal_pac_pos和bwa_refine_gapped放入結構體worker中。然后算法3中bwa_se_core_mt函數以worker作為線程函數創建線程并賦予每個線程一個線程號tid。根據線程的數量,預先確定讀入的reads的個數,將這些reads平分交給各個線程去處理,并且每個線程啟動時都帶有支持對每個reads子集進行一系列計算的數據結構。如算法2的1~10行,第2~9行將讀入的seqs按照線程數劃分,分給線程tid,然后第10行各個線程使用各自得到的seqs去執行bwa_aln2seq_core。bwa_cal_pac_pos和bwa_refine_gapped內部也是按照相同的方法劃分數據然后進行多線程處理的。
算法2 worker
輸入:work中各函數的參數,tid。
1 for i = 0 to n_seqs
2" #ifdef" HAVE_PTHREAD
3" //如果是多線程
4" if (i%n_threads != tid)
5"" //將讀入的n條seqs均勻劃分給各個線程,
6"" //每個線程只處理相應的seqs
7"" continue
8" end if
9"" end if
10 "bwa_aln2seq_core(tid,seqs+i);
11 end for
12 bwa_cal_pac_pos();
13 bwa_refine_gapped();
算法3 bwa_se_core_mt
輸入:.sai文件、reads。
輸出:.sam文件。
1 for i = 0 to number of threads
2 "if (bwa_read_seq() !=0)
3 ""pthread_create(worker,tid);//創建線程
4 "end if
5 end for
6 for i = 0 to number of threads
7 "pthread_join();//線程等待
8 end for
9 for i = 0 to n_seqs-1
10 "bwa_print_sam1();//將結果寫入.sam文件中
11 end for
在多線程的處理機制中,由于BWA的實現,存在兩個數據依賴點。第一個數據依賴點是BWT索引的訪問。由于比對必須引用reference序列的BWT索引,所以必須在啟動每個線程之前訪問并保留reference序列信息。之后存儲在內存中的BWT索引可以在線程之間共享。第二個數據依賴點是以SAM文件格式記錄計算結果,由于SAM文件reads的結果是有順序的,所以先將每個結果保留在內存中,直到所有線程完成它們的功能,再在最后階段按順序將它們寫入SAM文件格式[6]。
對于多線程改進,與BWA-MT和PBWA[6,29]不同的是,本文在此之前已經將SAMSE步驟的時間大大降低,因此對于一些reads數比較少的序列,多線程本身的開銷有可能會超過多線程所帶來的改進。
3 實驗結果與分析
3.1 實驗數據
實驗所使用的參考基因組為最新的人類數據集GRCh38.p14,待比對序列使用了從SRA數據庫中下載的不同長度的reads序列,分別是35 bp的SRR6889465、50 bp的SRR10067255、75 bp的SRR26828225和100 bp的SRR19540612。將每個序列均截取前一百萬、兩百萬、五百萬和一千萬條reads保存為新的fastq格式文件作為實驗數據。
值得一提的是,上面四組實驗數據都是來自與人類基因相關的基因。除此之外,還選擇了與人類基因無關的50 bp的SRR26561459和75 bp的SRR26152340來作為對照實驗。為了消除緩存引起的文件讀寫速度的差別,每次在進行實驗前會清空頁面緩存(PageCache)、目錄項和inode。
3.2 實驗平臺
本文所使用的CPU是英特爾酷睿i7-12700 處理器,共有12個內核,內存為32 GB。基于Ubuntu 20.04.2系統環境下的gcc 9.4.0完成實驗,使用的BWA版本為最新的0.7.17版本,所使用的多線程為POSIX中的pthread 2.31。
3.3 實驗結果
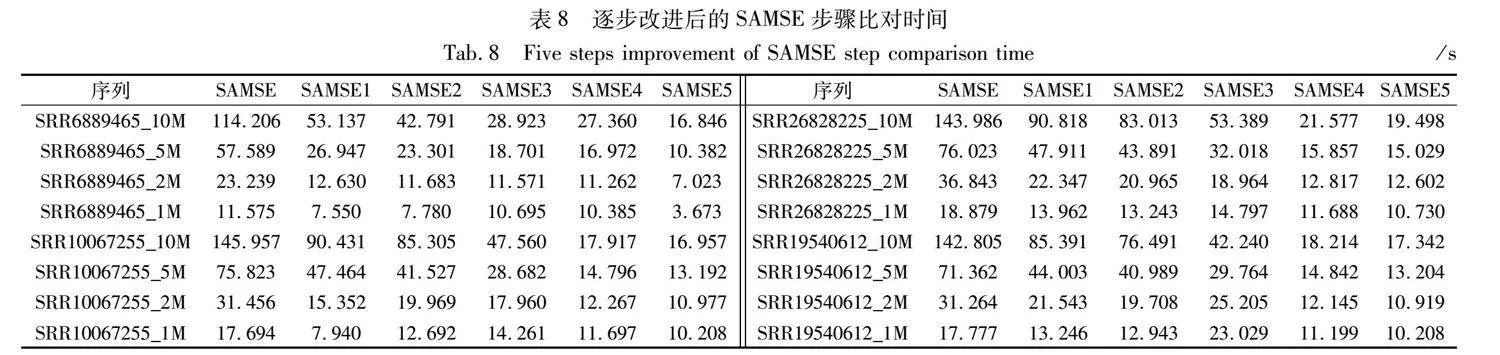
本文在BWA子算法SAMSE的基礎上,依次做了如下優化:去除SAMSE步驟中讀取SA數據的冗余、去除SAMSE步驟中讀取參考序列的冗余、選擇新的SA_INTERVAL參數、多線程優化和選擇新的OCC_INTERVAL參數。每次優化均在上次的基礎上進行,按順序將這五種優化依次命名為samse1、samse2、samse3、samse4和samse5,另外將原始SAMSE的實現命名為samse。下面將依次報告每次優化相較于前序優化的實驗。
3.3.1 SAMSE流程優化的實驗
表8展示了BWA的SAMSE步驟的實現samse逐步進行五次改進分別對不同的序列的比對時間,其中第一行為原始samse和其他五種版本的samse,第一列代表了不同序列的前一百萬、兩百萬、五百萬和一千萬條reads。其中2~5行(左)表示35 bp的序列SRR6889465的比對時間,6~9行(左)代表50 bp的SRR10067255的比對時間,1~4行(右)代表75 bp的SRR26828225的比對時間,5~8行(右)代表100 bp的SRR19540612的比對時間。從表8的第2、3列(左、右)可以看出消除讀入SA數據的冗余后,35 bp的序列的情況下,速度是samse的1.42~1.82倍,平均1.64倍;50 bp的序列的情況下,速度是samse的1.35~1.61倍,平均1.51倍;75 bp的序列的情況下,速度是samse的1.35~1.59倍,平均1.54倍;100 bp的序列的情況下,速度是samse的1.34~1.67倍,平均1.52倍。總的來看是1.34~1.82倍,平均1.55倍。并且reads數越多,改進就越大,在reads數為10 M的數據上,samse1是samse速度的1.59~1.82倍,平均為1.68倍。表8的第4列(左、右)展示的是在samse1基礎上又消除了讀入參考序列冗余的samse2的對比時間,與第3列(左、右)作對比可以看出4組實驗中,35 bp的序列的情況下,samse2速度是samse1的1.02~1.27倍,平均1.15倍;50 bp的序列的情況下,samse2速度是samse1的1.03~1.14倍,平均1.07倍;75 bp的序列的情況下,samse2速度是samse1的1.05~1.09倍,平均1.08倍;100 bp的序列的情況下,samse2速度是samse1的1.02~1.12倍,平均1.08倍。總的來看速度提升為1.02~1.27倍,平均1.10倍。在reads數為10 M的數據上,samse2是samse1速度的1.09~1.27倍,平均為3.78倍。samse2是samse速度的1.71~2.15倍,平均為1.87倍。而且與samse1類似,reads數越多,改進就越大。
3.3.2 SA_INTERVAL參數選擇的實驗
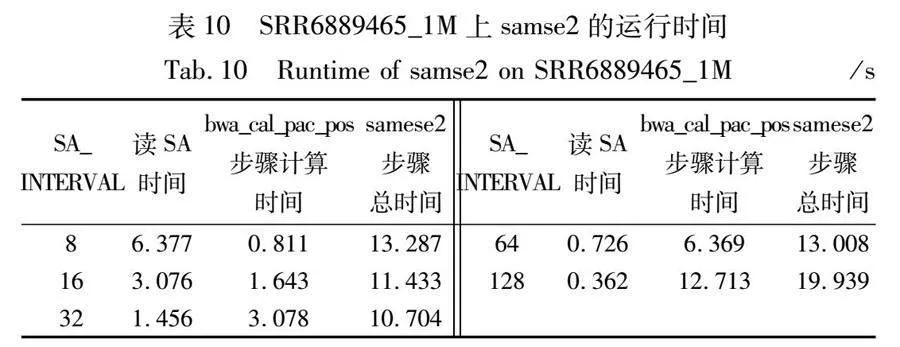
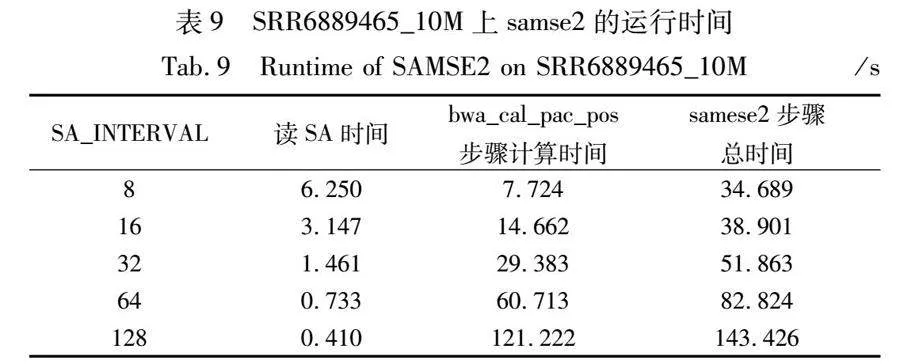
如表9所示是SRR6889465的前一千萬條reads組成的數據集的samse2時間隨SA_INTERVAL值在8~128的時間變化統計。通過實驗發現在SA_INTERVAL=8時,samse2的時間最短。對SAMSE的時間進行分析,發現受參數影響的時間主要是讀取SA的時間和bwa_cal_pac_pos步驟的計算時間。其中讀取SA的時間隨SA_INTERVAL的增大而減少,bwa_cal_pac_pos步驟的時間隨SA_INTERVAL的增大而增大。
進一步分析可知,讀取SA的時間是由參考序列的索引決定的,所以當參考基因組不變時,與任何reads在同一SA_INTERVAL的條件下比對時讀取SA的時間都是一樣的。而在做基因組研究時,一般都是用不同的reads來與某個參考基因組進行比較。因此選擇SA_INTERVAL的判斷依據是bwa_cal_pac_pos的計算量。
如表10所示是SRR6889465前一百萬條reads組成的數據集的比對時間統計,可以看出由于reads數減少了,bwa_cal_pac_pos步驟的計算時間也下降了,而是和讀取SA的時間屬于同一量級,所以綜合兩者來看SA_INTERVAL=32時比對速度最快,也可以看出reads的個數是影響bwa_cal_pac_pos計算時間的因素之一。
綜上所述,如果bwa_cal_pac_pos計算時間遠大于讀取SA時間的fastq文件,推薦選較小的SA_INTERVAL;如果bwa_cal_pac_pos計算時間遠小于讀取SA時間的fastq文件,推薦選較大的SA_INTERVAL;而當兩者時間是同一量級時,則推薦選擇默認參數32。而現代測序技術產生的數據一般都非常龐大,bwa_cal_pac_pos計算時間也會遠大于讀取SA的時間,因此SA_INTERVAL選擇8或以下。從4.5節的討論可知,選擇更小的SA_INTERVAL會導致內存消耗過大從而影響比對效率,因此結合本文實驗的硬件條件下選擇了8。實際上SA_INTERVAL的選擇應該更加靈活,筆者在文獻[30]提出了SA_INTERVAL的自動選擇模型。
表8的第5列(左、右)所示的是基于samse2版本選擇了新的SA_INTERVAL值(等于8)的samse3在四組數據上的運行時間。在35 bp的數據上,samse3的比對速度是samse2的0.84~1.49倍,平均1.17倍;50 bp的數據的情況為,samse3的比對速度是samse2的0.89~1.79倍,平均1.31倍;75 bp的數據的情況為,samse3的比對速度是samse2的0.90~1.56倍,平均1.23倍;100 bp的數據的情況為,samse3的比對速度是samse2的0.56~1.81倍,平均1.13倍。總的來看速度提升為0.56~1.81倍,平均1.21倍。特別地,在reads數為10 M的數據上,samse3是samse2速度的1.49~1.81倍,平均為1.66倍。samse3是samse速度的2.70~3.38倍,平均為3.12倍。
容易知道四組數據在reads數為一百萬和兩百萬的情況下之所以速度下降,是因為reads數的減少會讓bwa_cal_pac_pos函數的運行時間也會隨之下降,所以選擇小的SA_INTERVAL對bwa_cal_pac_pos的計算時間的改進被增加的讀時間超過了,在reads數比較小的情況下比對速度下降。
3.3.3 SAMSE的多線程實現
表8的第6列(左、右)代表的是基于samse3的基礎上加入了多線程的samse4的比較,其中35 bp的序列的情況下,samse4速度是samse3的1.21~1.93倍,平均1.54倍;50 bp的情況下samse4速度是samse3的1.22~2.65倍,平均1.82倍;75 bp的序列的情況下,samse4速度是samse3的1.27~2.47倍,平均1.81倍;100 bp的序列的情況下,samse4速度是samse3的2.01~2.32倍,平均2.11倍。總的來看速度提升為1.21~2.65倍,平均1.82倍。特別地,在reads數為10M的數據上,samse4是samse3速度的1.93~2.65倍,平均為2.34倍。samse4是samse速度的6.67~8.15倍,平均為7.34倍。
3.3.4 OCC_INTERVAL參數的影響實驗
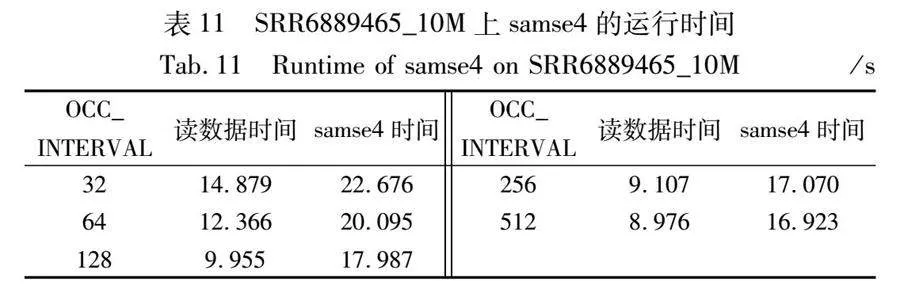
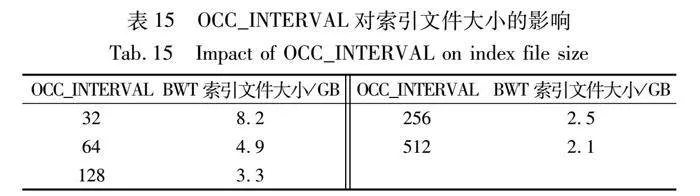
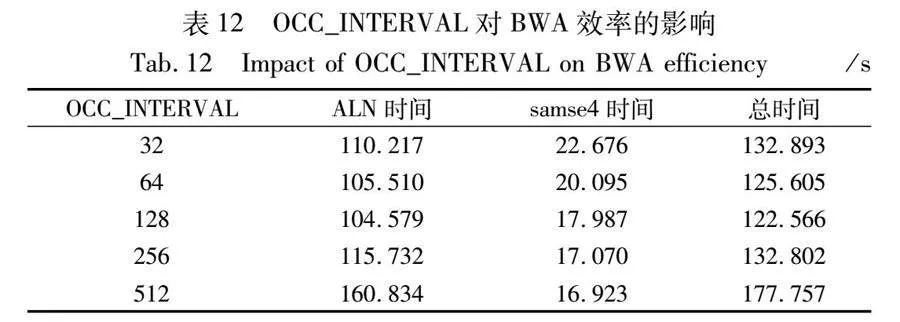
表11展示的是SRR6889465前一千萬條reads選擇不同OCC_INTERVAL的運行時間,可以看出OCC_INTERVAL越大,samse4的運行效率就越快。經過對SAMSE步驟分析,OCC_INTERVAL影響SAMSE運行效率主要是因為SAMSE步驟在讀入BWT的時候,會一起讀取OCC索引,所以OCC_INTERVAL選擇得越大,讀取的OCC索引就越少,花費的時間也越少,OCC_INTERVAL可以選擇較大的數值。
表8的第7列(左、右)顯示了在samse4基礎上選擇了512為新的OCC_INTERVAL samse5的比對速度。其中35 bp的序列的情況下,samse5速度是samse4的1.06~1.12倍,平均1.10倍;50 bp的序列的情況下,samse5速度是samse4的1.06~1.15倍,平均1.11倍;75 bp的序列的情況下samse5速度是samse4的1.02~1.11倍,平均1.07倍;100 bp的序列的情況下,samse5速度是samse4的1.05~1.12倍,平均1.10倍。總的來看速度提升為1.02~1.15倍,平均1.10倍。特別地,在reads數為10 M的數據上,samse5是samse4速度的1.05~1.12倍,平均為1.11倍。samse5是samse速度的7.11~8.61倍,平均為7.84倍。
但是和SA_INTERVAL不同,OCC_INTEVAL也會影響到ALN步驟的效率。表12展示了對SRR6889465前一千萬條reads改變OCC_INTERVAL后ALN和SAMSE步驟計算時間的變化。從目前的實驗來看,如果考慮BWA總的計算時間,OCC_INTERVAL的最佳選擇還是默認參數128。至此本文將包含samse4版本的BWA重新命名為BWA*。
3.3.5 BWA*與BWA的對比
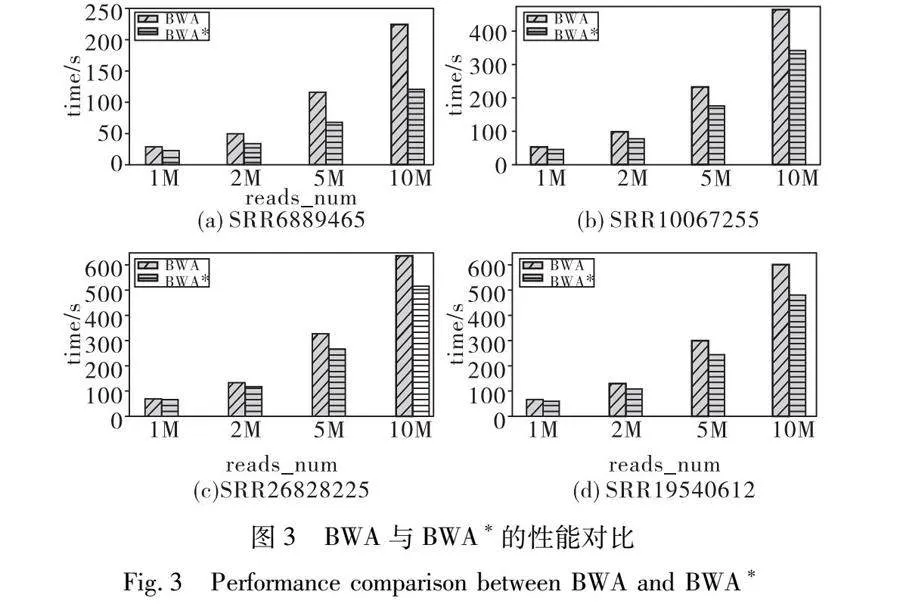
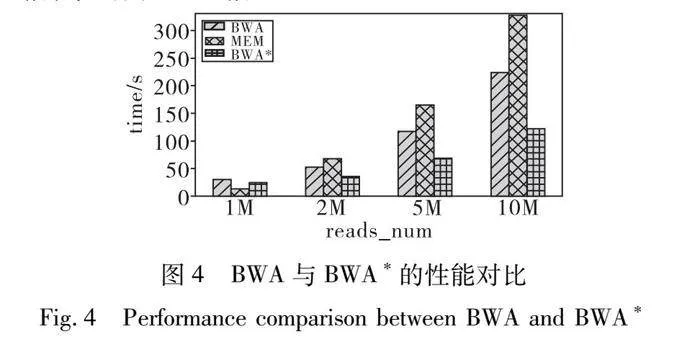
圖3是BWA和samse4版本的BWA*在4線程時,SAMSE步驟加上ALN步驟的總比對速度的比較。在35 bp時,BWA*的比對速度是BWA的1.22~1.84倍,平均1.56倍;在50 bp時,BWA*的比對速度是BWA的1.06~1.25倍,平均1.17倍;在75 bp時,BWA*的比對速度是BWA的1.09~1.25倍,平均1.19倍;在100 bp時,BWA*的比對速度是BWA的1.14~1.37倍,平均1.27倍;總體來看BWA*是BWA的1.09~1.84倍,平均1.30倍。特別地,在reads數為10M的數據上,BWA*是BWA速度的1.25~1.70倍,平均1.47倍。
此外,如圖4所示,是在35 bp時BWA*、BWA和BWA-MEM的性能對比。容易看出只有在reads為一百萬條的情況下,BWA-MEM更快,分別是BWA和BWA*的1.81和1.69倍。而在兩百萬、五百萬和一千萬條的時候,則是BWA和BWA*更快,BWA在這三組數據上是BWA-MEM的1.31、1.42和1.46倍,BWA*在這三組數據上是BWA-MEM的1.91、2.44和27倍。BWA*是BWA的1.22~1.84倍,平均為1.56倍;BWA*在reads大于兩百萬條的數據上是BWA-MEM的1.91~2.67倍,平均為2.34倍。
3.4 read與參考基因組的相似性對實驗結果影響的討論
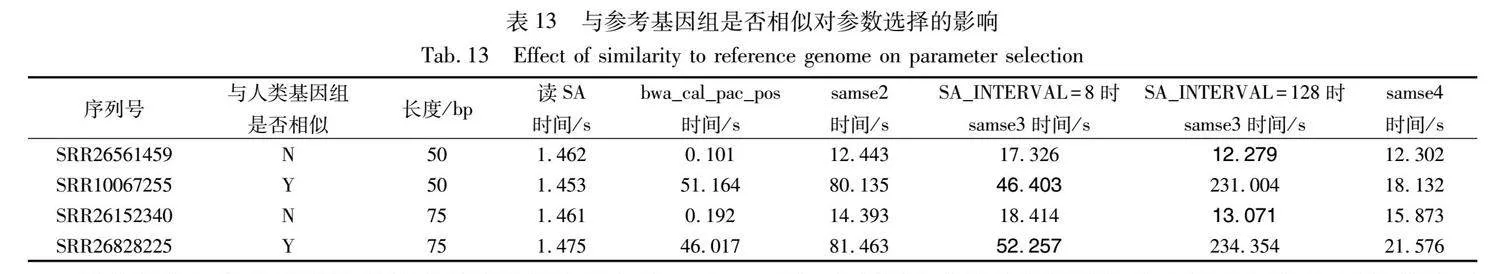
為了實驗的公平性和客觀性,使用了與人類基因完全無關的SRR26561459和SRR26152340以及與人類基因組相似的SRR10067255和SRR26828225的前一千萬條reads與人類基因組進行了比對實驗,結果如表13所示。其中第7和8列中加粗的比對時間,是該行數據選擇了其最佳SA_INTERVAL參數后的比對時間。最后一列samse4中表示的則是各數據在選擇了其最佳SA_INTERVAL參數后,加入了多線程改進的比對時間。與人類基因無關的SRR26561459和SRR26152340都是在SA_INTERVAL=128時速度最快,速度分別是默認參數時的1.01和1.10倍;與人類基因相似的SRR10067255和SRR26828225都是在SA_INTERVAL=8時速度最快,速度分別是默認參數時的1.73和1.56倍。
從表13可以看出reads長度相同時,與參考基因組相似數據的bwa_cal_pac_pos步驟的計算時間要遠遠大于與參考基因組不相似的數據。這是因為如果數據中的reads都沒有比對到參考基因組上,那么SAMSE步驟的一些結果信息并不會被計算,包括bwa_cal_pac_pos步驟,因為沒有比對上,所以也就沒有比對位置可以還原;如果數據中的reads都能比對到參考基因組上,那么就會執行更多次bwa_cal_pac_pos。因此read序列與參考基因組相似與否也是影響SA_INTERVAL選擇的重要因素,如果相似,bwa_cal_pac_pos計算時間會更多,則更傾向于選擇小的SA_INTERVAL;反之,如果不相似,bwa_cal_pac_pos計算時間會更少,更傾向于選擇大的SA_INTERVAL。
表13還展示了四組數據與人類基因組在多線程下的samse4比對時間,可以看出SRR26561459和SRR26152340兩組不相似的數據在加入多線程后速度不但沒有增長反而下降了。而與參考基因組相似的SRR10067255和SRR26828225則在加入多線程后,速度得到了很大提升,分別是原來速度的2.56和2.42倍。
這是因為與參考基因組相似度高的數據比對上的reads比較多,所以SAMSE步驟的計算量會更大。相反,與參考基因組相似度低的數據比對上的reads很少,因此SAMSE步驟的計算量很小,甚至多線程的開銷超過了所得到的提升,所以速度反而下降了。因此,數據中的reads與參考基因組相似與否也是判斷SAMSE是否需要多線程的重要依據。
綜上所述,reads與參考基因組相似度對于SA_INTERVAL的選擇和reads是否適合多線程具有重要影響。對于SA_INTERVAL的選擇,一是要看reads是否與參考基因組相似,比如可以通過兩基因來源的生物的種群來判斷,相似則傾向于選擇比較小的SA_INTERVAL,反之則選擇比較大的SA_INTERVAL;二是看reads的個數,reads數比較多則選擇比較小的SA_INTERVAL,反之則選擇比較大的SA_INTERVAL。而對于比對是否要使用多線程,與參考基因組相似度高的reads使用多線程比對的速度容易得到提升,相似度低的則不推薦使用多線程。
3.5 參數對內存占用影響的討論
3.3節的實驗展示了SA_INTERVAL和OCC_INTERVAL兩個參數對BWA計算效率的影響。但除了運行時間外,參數值對內存占用的影響也是一個需要考慮的因素。
對于序列比對的內存占用大小,對于ALN主要分為以下幾個部分:參考序列、參考序列的索引、reads和比對產生的結果信息,其中主要部分是參考序列和索引部分,索引部分具體分成參考序列的BWT、SA和OCC。以人類基因組為例,壓縮后的二進制形式參考序列需要0.8 GB內存;索引部分中,參考序列的BWT,包括正向和反向BWT需要1.6 GB內存。對于其他索引,在SA_INTERVAL和OCC_INTERVAL都是默認參數的情況下,SA部分需要1.6 GB內存,OCC部分需要1.6 GB內存。reads由于是分批讀進去的,結果信息也是分批生成并寫入結果文件的,加上一些其他開銷,總共只有200~300 MB。
BWA算法有ALN和SAMSE兩個部分組成。其中ALN步驟所需要的內存主要有BWT、SA索引、reads以及其他開銷,默認參數下內存開銷為3.4 GB左右。SAMSE步驟所需要的內存開銷有BWT、SA索引、OCC索引、參考序列和reads等,默認參數下總內存開銷為5.7 GB左右。
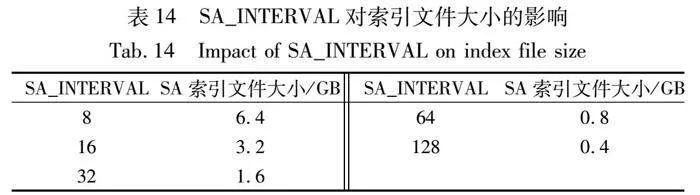
表14、15顯示了改變SA_INTERVAL和OCC_INTERVAL的值對索引文件大小的影響,索引文件的大小會直接影響內存占用的多少。值得一提的是,索引文件包括.amb、.ann、.bwt、.sa和.pac文件,其中.amb和.ann都是關于參考基因組的注釋信息,大小只有幾十KB,.bwt文件主要包括的是OCC索引和參考序列的BWT,.sa文件包含的是SA索引,.pac文件包含的壓縮后的參考序列。根據3.2節,可知BWA的SA和OCC索引是部分保存的,因此可以改變SA_INTERVAL和OCC_INTERVAL來改變索引的大小。從兩表中可以看出隨著SA_INTERVAL和OCC_INTERVAL值的增加,.bwt和.sa索引文件的大小隨之下降從而降低內存的占用,反之則會增加內存的占用。如果兩個參數的值同時取表中的最小值,SAMSE步驟至少需要15 GB以上的內存,這可能會導致配置較低的機器內存資源不足的問題,因此實際計算中參數值的選擇還需要考慮到計算平臺的內存大小,筆者在文獻[30]提出了對基于BWT的序列比對算法的內存峰值估計模型,并進行了更細致的分析如表14、15所示。
3.6 BWA*在古代DNA比對的應用
在古代DNA領域,使用的最多的序列比對算法是BWA和BWA-MEM[15],主要是因為BWA在比對古代DNA時精度較高,而BWA-MEM相對于BWA精度略低,但是速度較快,所以古代研究者經常在選擇比對工具方面產生分歧[15,16]。
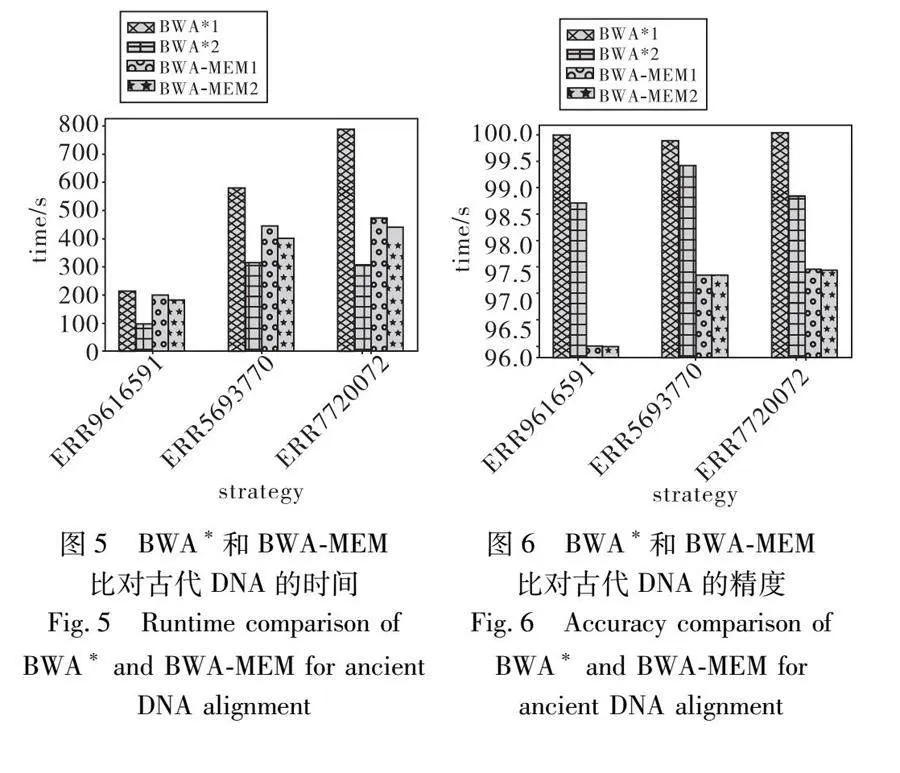
使用改進后的BWA*和BWA-MEM在默認SA_INTERVAL和OCC_INTERVAL下使用表16中古代DNA比對時常用的幾組參數對古代DNA進行比對,得到BWA*和BWA-MEM在比對古代DNA時的時間和精度對比,如圖5、6所示。
從圖5可以看出BWA*在參數選擇BWA*2時比對古代DNA時的速度已經超越了BWA-MEM,而且由于BWA*在提升速度的同時并未影響其精度,可以在圖6中看出BWA*在比對古代DNA時仍保持了其高精度的特點。
4 結束語
本文對序列比對算法BWA,主要是其SAMSE步驟,進行了流程優化、參數值調整和多線程加速等一系列優化。實驗表明,當使用與人類參考基因組相似的reads時,算法流程優化和多線程加速對不同長度和個數的reads都有提升,而使用新的SA_INTERVAL默認參數值對reads數較多的數據有明顯提升。 使用新的OCC_INTERVAL默認參數值,對不同長度和個數的reads,SAMSE的速度都有提升,但ALN步驟的速度有所下降。值得注意的是BWA*在古代DNA的比對中的效果令人欣喜,保持高精度的同時速度超越了BWA-MEM。下一步工作,筆者將會考慮將BWA*中的優化方法,包括對參數的自動調優,應用到其他以BWT作為索引方式的序列比對算法。
參考文獻:
[1]Satam H, Joshi K, Mangrolia U,et al. Next-generation sequencing technology: current trends and advancements [J]. Biology, 2023, 12(7): 997.
[2]Pennisi E. Upstart DNA sequencers could be a ‘game changer’[J]. Science, 2022, 376(6599): 1257-1258.
[3]Gorzynski J E, Goenka S D, Shafin K,et al. Ultrarapid nanopore genome sequencing in a critical care setting [J]. New England Journal of Medicine, 2022, 386(7): 700-702.
[4]Alser M, Lindegger J, Firtina C,et al. From molecules to genomic variations: accelerating genome analysis via intelligent algorithms and architectures[J]. Computational and Structural Biotechnology Journal, 2022, 20: 4579-4599.
[5]溫華銘, 徐云, 楊金寶. 串聯重復序列比對的位置篩選方法 [J]. 計算機應用研究, 2024, 41(7): 2160-2164. (Wen Hua-ming, Xu Yun, Yang Jinbao. Position filtering method for tandem repeat sequence alignment [J]. Application Research of Compu-ters, 2024, 41(7): 2160-2164.)
[6]Jo H, Koh G. Faster single-end alignment generation utilizing multi-thread for BWA [J]. Bio-Medical Materials and Engineering, 2015, 26(S1): S1791-S1796.
[7]崔英博, 黃春, 唐滔, 等. 基因組大數據變異檢測算法的并行優化[J]. 大數據, 2020, 6(5): 16-28. (Cui Yingbo, Huang Chun, Tang Tao, et al. Parallel optimization of variation detection algorithms for large-scale genome data[J]. Big Data Research, 2020, 6(5): 16-28.)
[8]Alser M, Rotman J, Deshpande D,et al. Technology dictates algorithms: recent developments in read alignment [J]. Genome Bio-logy, 2021, 22(1): 249.
[9]Xin Yao, Liu Benben, Min Biao, et al. Parallel architecture for DNA sequence inexact matching with Burrows-Wheeler transform[J]. Microelectronics Journal, 2013, 44(8): 670-682.
[10]Burrows M. A block-sorting lossless data compression algorithm, 124[R]. Palo Alto: Systems Research Center, 1994.
[11]Langmead B. Introduction to the Burrows-Wheeler transform and FM-index [EB/OL].(2013-11-24) [2023-12-20]. https://www.cs.jhu.edu/~langmea/resources/lecture_notes/bwt_and_fm_index.pdf.
[12]Li Heng, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform [J]. Bioinformatics, 2009, 25(14): 1754-1760.
[13]Al Kawam A, Khatri S, Datta A. A survey of software and hardware approaches to performing read alignment in next generation sequencing[J]. IEEE/ACM Trans on Computational Biology and Bioinformatics, 2016, 14(6): 1202-1213.
[14]Li Heng. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM [EB/OL]. (2013-05-26). https://arxiv.org/abs/1303.3997.
[15]Sarmento C, Guimares S, Kln G M,et al. A study on Burrows-Wheeler aligner’s performance optimization for ancient DNA mapping[C]// Proc of the 15th International Conference on Practical Applications of Computational Biology amp; Bioinformatics. Cham: Springer, 2022: 105-114.
[16]Oliva A, Tobler R, Cooper A,et al. Systematic benchmark of ancient DNA read mapping [J]. Briefings in Bioinformatics, 2021, 22(5): bbab076.
[17]宋光捷, 蔡大偉, 朱存世, 等. 古DNA揭示中國西北地區史前人類對野生動物資源的利用[J/OL]. 人類學學報. (2024-05-17). https://doi.org/10.16359/j.1000-3193/AAS.2024.0031. (Song Guangjie, Cai Dawei, Zhu Cunshi, et al. Ancient DNA reveals the utilization of wild animal resources by prehistoric humans in Northwest China[J/OL]. Acta Anthropologica Sinica. (2024-05-17). https://doi.org/10.16359/j.1000-3193/AAS.2024.0031.)
[18]Wang Zihang, Cui Yingbo, Peng Shaoliang, et al. MinimapR: a pa-rallel alignment tool for the analysis of large-scale third-generation sequencing data[J]. Computational Biology and Chemistry, 2022, 99: 107735.
[19]Li H. BWA manual sourceforge [EB/OL].(2023-12-20). http://bio-bwa.sourceforge.net/bwa.shtml.
[20]Liu Yuansheng, Shen Xiangzhen, Gong Yongshun, et al. Sequence alignment/map format: a comprehensive review of approaches and applications[J]. Briefings in Bioinformatics, 2023, 24(5): bbad320.
[21]Rhoads A, Au K F. PacBio sequencing and its applications [J]. Genomics, Proteomics amp; Bioinformatics, 2015, 13(5): 278-289.
[22]Li Heng. Minimap2: pairwise alignment for nucleotide sequences[J]. Bioinformatics, 2018, 34(18): 3094-3100.
[23]Pham-Quoc C, Kieu-Do B, Thinh T N. A high-performance FPGA-based BWA-MEM DNA sequence alignment[J]. Concurrency and Computation: Practice and Experience, 2021, 33(2): e5328.
[24]Vasimuddin M, Misra S, Li H,et al. Efficient architecture-aware acceleration of BWA-MEM for multicore systems[C]// Proc of IEEE International Parallel and Distributed Processing Symposium. Piscata-way, NJ: IEEE Press, 2019: 314-324.
[25]Pham M, Tu Yicheng, Lyu Xiaoyi. Accelerating BWA-MEM read mapping on GPUs[C]// Proc of the 37th International Conference on Supercomputing. New York: ACM Press, 2023: 155-166.
[26]Song Y J, Cho D H. Local alignment of DNA sequence based on deep reinforcement learning[J]. IEEE Open Journal of Engineering in Medicine and Biology, 2021, 2: 170-178.
[27]Roodi M, Lak Z, Moshovos A. BWA-MEM performance:suffix array storage size[C]// Proc of IEEE EMBS International Conference on Biomedical amp; Health Informatics. Piscataway, NJ: IEEE Press, 2019: 1-4.
[28]Ferragina P, Manzini G. Opportunistic data structures with applications[C]// Proc of the 41st Annual Symposium on Foundations of Computer Science.Piscataway, NJ: IEEE Press, 2000: 390-398.
[29]Jo H. Multi-threading the generation of Burrows-Wheeler alignment[J]. Genetics and Molecular Research, 2016,15(2): 27323088.
[30]Hu Shuang, Chen Changbo. Performance models for sequence alignment algorithms based on Burrows-Wheeler transform[C]// Proc of the 16th International Conference on Bioinformatics Biomedical Technology. New York: ACM Press, 2024.
