生酮飲食治療CDKL5基因突變所致嬰兒痙攣癥1例報告并文獻復習
2024-05-09 12:01:22王春雨王琳琳南在元劉爽張哲
中國醫藥指南 2024年12期
關鍵詞:癲癇
王春雨,王琳琳,南在元,劉爽,張哲
首都醫科大學附屬北京兒童醫院黑龍江醫院(哈爾濱醫科大學附屬第六醫院),哈爾濱 150010
CDKL5 基因全稱為細胞周期依賴蛋白激酶樣蛋白5(cyclin-dependent kinase-like 5,CDKL5),是X染色體連鎖基因,定位于Xp22.13,編碼絲氨酸-蘇氨酸蛋白激酶,在大腦中廣泛分布,是大腦正常發育的必需蛋白。CDKL5 蛋白在大腦中行使激酶功能,它可以通過磷酸化下游底物,從而改變其他蛋白的活性,參與神經發育及神經功能的發揮[1]。多項研究顯示,CDKL5 基因突變涵蓋的表型譜廣泛,臨床癥狀與嬰兒痙攣癥、Rett 綜合征、Angelman 綜合征、自閉癥等存在交叉部分,多數表現為重度的發育遲緩、嬰幼兒早發性難治性癲癇、重度的肌張力障礙、類Rett 綜合征、類Angelman 綜合征等,稱為CDKL5基因相關早發性癲癇性腦病,又稱CDKL5 基因相關疾病[2]。癲癇發作通常是CDKL5 基因相關疾病的核心癥狀,患兒通常表現出三個階段的癲癇發作:Ⅰ期是早期癲癇,多表現為強直發作(發病1~10 周),腦電圖正常。Ⅱ期是發育性癲癇,通常伴有嬰兒痙攣,發作間期腦電圖高度失律。Ⅲ期多表現為晚期,局灶性發作、肌陣攣發作和陣攣性發作,也可為強直發作及不典型失神發作等[3],均逐漸發展為難治性癲癇,同時可合并有嚴重精神運動發育遲滯,如豎頭不穩、不會說話等,常有肌張力低下的表現。使用激素聯合多種抗癲癇發作藥物治療欠佳的CDKL5 基因突變所致嬰兒痙攣癥1 例,應用生酮飲食(ketogenic diet,KD)治療后,患兒在癲癇發作、運動及智力方面均有一定改善,特總結如下。
1 病例報告
1.1 基本情況 患兒男,2 月16 天,主因“間斷抽搐20 余日”于2020 年7 月入院。患兒出生后1 月24 天起出現抽搐發作,表現為雙目凝視、雙拳緊握、頭后仰、雙側肢體抖動,以右側為著,每次發作持續數秒鐘至1 分鐘自行緩解,每日均有發作。外院給予左乙拉西坦口服無明顯好轉故來我院。住院后第4 天抽搐發作形式改變,表現為單下或成串的短暫點頭伴四肢屈曲或伸展樣強直,發作多于睡醒后短時間內出現,伴大哭,成串發作時,每串5~20 下,3~4 串/d。患兒G2P1,正常順產,無生后窒息史,出生時無臍帶繞頸,羊水及胎盤情況正常。其母孕第1 胎為死胎,母親否認疾病史,患兒直系親屬否認癲癇病史。患兒生后混合喂養,進乳良好,體重增長良好。
1.2 體格檢查 意識清楚,精神可,呼吸平穩,面色正常,頭圍:37 cm,體重:6.5 kg,前囟平坦,為1.0 cm×1.0 cm,周身未見皮疹及出血點,淺表淋巴結未觸及腫大。咽部正常,肺部聽診未聞及干濕啰音,心臟聽診節律規整,無心律失常,心率126 次/min,未聞及收縮期和舒張期雜音。腹部平坦、柔軟,肝脾肋下未觸及,脊柱未見異常,四肢末梢溫暖。神經系統查體:意識清楚,精神可,雙球結膜無水腫,無顱神經癱瘓,四肢肌力Ⅴ級,肌張力無增高或減低,雙膝腱反射、跟腱反射正常引出,淺反射正常,雙巴氏征陰性,頸強陰性,克氏征、布什征陰性。
1.3 輔助檢查資料 視頻腦電圖:高度失律,監測到痙攣發作(圖1a,1b)。頭磁共振+腦功能成像:雙側額顳極腦外間隙略寬;顱腦DWI 未見明顯異常(圖2a,2b)。血常規、血生化、血氨、血乳酸等相關代謝指標:未見明顯異常。甲狀腺功能、甲狀旁腺素:未見異常。心臟超聲、腹部超聲、胸片:未見異常。血尿代謝篩查:陰性。染色體核型分析未見異常。經醫院倫理委員會通過,患兒父母即監護人簽署基因檢測知情同意書后,同時收集患兒及父母外周血樣本,委托第三方智因東方轉化醫學研究中心檢驗。患兒血液標本進行高通量測序,父母血液標本進行驗證。全基因組測序結果發現:患兒CDKL5基 因chrX:18638087,c.2376+1(IVS16)G >A,(NM_001323289),嵌合新生變異,其父母無此位點的突變,為致病變異(圖3a,3b,3c),為X 連鎖顯性遺傳。

圖1a 視頻腦電圖睡眠期高度失律

圖2a 頭顱磁共振水平面雙側額顳極腦外間隙略寬
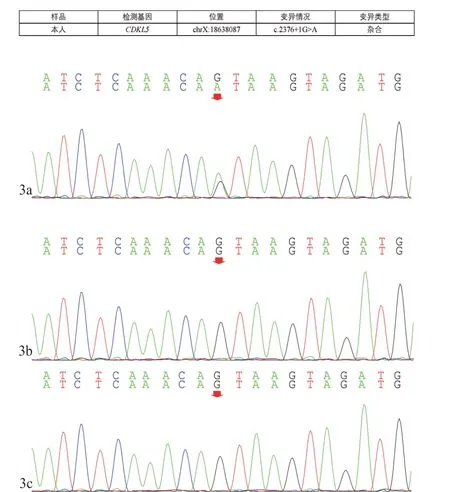
圖3 家系CDKL5測序峰圖
1.4 診斷、治療和隨訪 患者診斷:癲癇(局灶性發作、痙攣發作),嬰兒痙攣癥,CDKL5 相關發育性癲癇性腦病。治療:患兒診斷為嬰兒痙攣癥后,給予促腎上腺皮質激素(ACTH)12.5 IU 靜脈滴注2 周,減停左乙拉西坦換用托吡酯抗癲癇發作治療,發作有減少,但仍有發作,ACTH 加量為25 IU 靜脈滴注2 周,加用丙戊酸鈉聯合抗癲癇發作治療,ACTH 共應用4 周發作停止,腦電圖改善,序貫醋酸潑尼松口服,激素共12 周減停。1 個月后(6.5 月齡)患兒再次出現痙攣發作,5~6 串/d,5~15 下/串,且發作逐漸增多,加用氨己烯酸口服并逐漸加量,發作減少,但仍有每日3~4 串發作,并逐漸出現發育倒退。2 個月后(2021.01.05,患兒8.5 月齡)患兒仍不能獨坐,出現翻身困難,四肢肌張力減低,仍有痙攣發作,每日2~3 串。患兒完善血尿代謝篩查及基因檢測排除生酮飲食禁忌證后,開始給予生酮飲食治療。生酮飲食采用生酮奶,開始時脂肪與(碳水化合物+蛋白質)的質量比為2 ∶1,根據患兒耐受情況逐漸調整至3 ∶1~4 ∶1,使血酮維持在3~5 mmol/L,血糖維持在4~5 mmol/L,口服托吡酯、丙戊酸鈉、氨己烯酸抗癲癇發作。生酮飲食治療3 個月后,癲癇發作明顯減少,痙攣發作2~5 日1 次。堅持生酮飲食2 年,痙攣發作消失,運動發育進步,可翻身、獨坐,腦電圖改善,無高度失律(圖4a,4b)。但因患兒反復患“支氣管肺炎”及“重癥肺炎”,進食生酮奶及生酮食品時反復嗆奶及嘔吐,患兒體重減輕明顯,3 歲時(2023 年4 月)體重8 kg,家長難于堅持停用生酮飲食。停用生酮飲食后局灶性發作增加,每日4~5 次,每次持續數秒鐘到1 分鐘可自行緩解,未再有痙攣發作,目前口服丙戊酸鈉、托吡酯、拉莫三嗪抗癲癇發作。
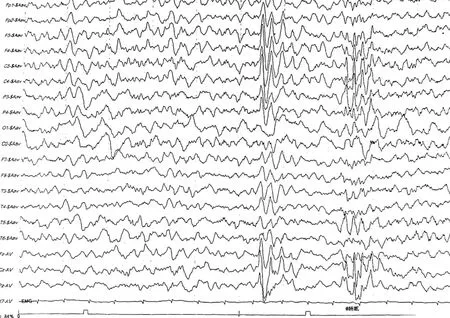
圖4a 視頻腦電圖生酮飲食治療后睡眠期
2 討論
CDKL5 基因相關疾病是由于CDKL5 基因突變所引起的一種罕見的神經發育性疾病,患者的臨床表現多樣,主要表現為早發性難治性癲癇,以及神經系統發育遲緩導致的認知、運動、語言和視覺功能障礙[1]。CDKL5 基因相關疾病在新生兒中的發病率為1 ∶60000 至1 ∶40000,且患兒多為女性,男性患者報道較少,往往有更嚴重的臨床表現[4]。CDKL5基因致病性變異所致癲癇主要為早發性癲癇,首發中位年齡為6 周(1 周至1.5 歲),90%的癲癇發作時間為生后3 個月內。癲癇最早被描述為三個階段:第一階段是早發性驚厥發作,盡管發作間期腦電圖正常;第二階段是癲癇性痙攣,高度失律;第三階段是進行性的難治性癲癇發作;近一半的患者服用三種或三種以上抗癲癇藥物,大多患者為藥物難治性癲癇[5],同時可合并有嚴重精神運動發育遲滯,如豎頭不穩、不會說話等,常有肌張力低下的表現。本例患兒癲癇發作首發年齡為1 月24 天,病初表現為局灶性發作,抗發作治療效果不理想,繼而出現痙攣發作,與國內外報道多在3 月內發病相一致。在ACTH 治療及規律的抗癲癇發作藥物治療下,有所好轉,停用激素后發作加重,多種抗癲癇發作藥物聯合應用難于控制,逐漸出現發育遲滯及肌張力減低。應用生酮飲食治療后發作控制,智力、運動有所改善。停用生酮飲食后再次發作增加。
生酮飲食(ketogenic diet,KD)對難治性癲癇有較好的療效,被越來越多地用于難治性癲癇的治療[6-7],KD 能夠減少難治性癲癇患兒的發作,改善認知、言語和行為等問題。KD 是一個脂肪高比例、碳水化合物低比例,蛋白質和其他營養素合適的配方飲食,治療兒童難治性癲癇已有十余年的歷史。可能的作用機制為:對哺乳動物雷帕霉素靶蛋白(mTOR)及免疫的調節作用,mTOR 是人類中參與調節細胞大小,生長,蛋白質合成,自噬和轉錄的蛋白質,KD 可通過抑制mTOR 活性來減輕免疫反應;另外糖是大腦的主要能量來源,KD 可通過減少葡萄糖對大腦的直接供應而減輕興奮度,以此來達到抗癲癇效果[8-9]。由生酮飲食提供的多不飽和脂肪酸可能激活過氧化物酶體增殖物激活受體,該受體調節抗炎、抗氧化和線粒體基因,從而增加能量儲備,穩定突觸功能和限制過度興奮。從20 世紀初生酮飲食就開始應用于藥物難治性癲癇患兒。目前生酮飲食已被證實在多種癲癇綜合征中療效顯著[10-12],并成為兒童難治性癲癇的重要治療方法。近年來,生酮飲食治療逐漸被應用到了嬰兒痙攣癥的治療中。
嬰兒痙攣癥是一種嬰兒期常見的難治性癲癇綜合征,主要特點為嬰兒期起病、頻繁的痙攣發作、腦電圖出現高度失律和智力發育障礙。3~7 個月發病者最多,發作時表現為兩臂前舉,頭和軀干前屈,似點頭擁抱狀;少數患兒可呈頭背后屈。患兒常成簇發作,思睡或剛醒時容易連續發生,發作時有時伴喊叫、哭吵或痛苦狀,發作間期腦電圖示不對稱、不同步、并伴有暴發抑制交替傾向的高幅慢波,雜以多灶性尖波、棘波或多棘波,即高度失律。嬰兒痙攣癥病因多種多樣,可分為產前因素、圍生期因素及產后因素三類[13]。其中遺傳因素為產前因素的一部分,此例患兒為CDKL5 基因chrX:18638087,c.2376+1(IVS16)G >A,嵌合新生變異所致的嬰兒痙攣癥。促腎上腺皮質激素、糖皮質激素和氨己烯酸被作為一線治療嬰兒痙攣癥的短期療法,但停用后易于復發。越來越多的研究報道生酮飲食較多的應用于嬰兒痙攣癥中。王竹巖[13]、胡春輝[14]等對25 例經抗癲癇治療失敗的嬰兒痙攣癥患兒進行生酮飲食治療,其中7例(28%)的患者達到無臨床發作,且均在治療1 個月內顯效。吳革菲等[15]通過對部分藥物治療失敗的嬰兒痙攣癥患兒給予生酮飲食治療達到無發作。來自國際CDKL5 基因相關疾病數據庫的數據顯示,生酮飲食可以降低該基因所致痙攣發作頻率[16-17]。Pires等人對17 例嬰兒痙攣癥患兒應用生酮飲食治療的前瞻性研究表明,開始1 個月后6 例患兒無發作(35%),13 例患兒減少>50%發作(76%)。5 例腦電圖顯示高度失律的患兒中,有4 例在1 個月內消失(80%)。有3 例無癲癇發作的患者有精神運動改善。在開始生酮飲食治療3 個月后,11 例(65%)患兒無癲癇發作[18]。Kayyali 等[19]的研究表明,開始生酮飲食后3 個月,20%患兒減少>90%發作,該數據在6 個月時為22%,12 個月時為35%。研究認為盡早使用生酮飲食治療,除可以控制癲癇發作,還可以避免大劑量應用激素帶來的不良反應及降低復發風險,同時可以改善腦電圖,改善患兒的認知[20]。
近年來,國內外關于生酮飲食治療兒童難治性癲癇患兒的研究[21-23]顯示,3、6、12 個月末保留率分別為62.8%~80.7%、42.0%~70.0%、24.3%~54.0%。隨著生酮飲食治療時間的延長保留率逐漸減低的原因主要為,在生酮飲食治療實施過程中,部分患兒因生酮飲食治療適應時間偏長或不良反應明顯,不能完成營養師規定飲食,血酮在短時間內難以達到較穩定的水平,其中主要的不良反應包括初期拒食、嘔吐,不能完成規定飲食,部分患兒治療早期血酮均值<2 mmol/L。另外,生酮飲食限制嚴格,部分患兒及其監護人依從性差,因此提高患兒監護人對該治療的認識,幫助其建立信心,提高患兒及其監護人在生酮飲食治療過程中的依從性,從而提高生酮飲食治療保留率,有助于改善生酮飲食治療兒童難治性癲癇的療效。本例患兒在治療過程中先后應用左乙拉西坦、ACTH、托吡酯、丙戊酸鈉、氨己烯酸、拉莫三嗪,癲癇曾一度有所控制,再次發作加重后應用生酮飲食后癲癇控制較好,且智力、運動有所改善。但由于生酮飲食患兒長期耐受欠佳,堅持2 年后停用,痙攣發作消失,局灶性發作再次出現。
綜上所述,CDKL5 相關癲癇性痙攣及其相關綜合征臨床表現嚴重程度與突變位點、突變形式等密切相關,預后與臨床表現嚴重程度密切相關,預后常不理想,在口服抗癲癇發作藥物控制發作的前提下,生酮飲食治療可減少發作、改善生活質量,癲癇癥狀控制后患兒智力發育可出現進步。未來還有待于更多的樣本量的研究。
猜你喜歡
中國民間療法(2021年5期)2021-06-09 09:21:04
中華養生保健(2020年2期)2020-11-16 00:49:00
解放軍醫學院學報(2020年12期)2020-03-29 05:11:46
中成藥(2017年6期)2017-06-13 07:30:35
飲食科學(2017年5期)2017-05-20 17:11:53
臨床醫藥文獻雜志(電子版)(2017年11期)2017-05-17 04:48:10
安徽醫科大學學報(2015年9期)2015-12-16 11:09:44
中國當代醫藥(2015年7期)2015-03-01 02:01:13
西南軍醫(2015年4期)2015-01-23 01:19:30
西部中醫藥(2014年6期)2014-03-11 16:07:47
