土壤/沉積物中植酸對砷、磷形態轉化和生物有效性影響綜述
2024-05-09 03:34:52吳博賢艾雯妍文思穎楊曉莉
生態與農村環境學報 2024年4期
吳博賢,艾雯妍,文思穎,楊曉莉,劉 雪①
(1.西南林業大學生態與環境學院,云南 昆明 650224;2.西南林業大學環境修復與健康研究院,云南 昆明 650224)
土壤是人類賴以生存的基礎,土壤質量直接影響作物食品安全與人類健康。工農業生產中廢污水直排導致我國土壤重金屬污染形勢嚴峻。砷(As)在環境中分布廣泛,富砷地下水和地區污水灌溉導致砷在局部土壤/沉積物中大量積累,危及環境質量安全、農作物食品安全及底棲動物生存[1]。我國土壤砷含量均值為11.2 mg·kg-1,約為全球土壤砷含量均值(6 mg·kg-1)的2倍[2]。湖南郴州市柿竹園礦區土壤砷含量高達360~1 053 mg·kg-1[3],廣西、貴州、廣東等地亦出現土壤和水體砷污染現象[4]。砷污染具有隱蔽性、長期性和不可逆性等特點。據ALLAWAY[5]估算,進入土壤中的砷(6 mg·kg-1)通過植物吸收去除需約100 a。因此,土壤砷污染治理難度大、周期長。土壤中砷過量會導致農作物減產,沉積物中砷亦為潛在污染源,具有二次釋放風險,且可在生物體內積累,并通過食物鏈傳遞、富集進入人體,對人體健康構成威脅。
磷(P)是植物生長必需的大量營養元素。土壤/沉積物中磷與鐵(Fe)、鋁(Al)、鈣(Ca)、有機質等形成結合形態,在物理、化學、生物等作用下,通過解吸、溶解、還原等過程釋放進入土壤溶液和沉積物中,轉化為生物可利用態,成為誘發土壤農業面源污染和湖泊富營養化的重要因子[6]。土壤/沉積物中磷包括無機磷(IP)和有機磷(OP),溶解態無機磷(DIP)可被植物直接吸收,有機磷則需經礦化水解轉化為無機磷才可被植物吸收利用。有機磷占土壤總磷的40%~95%,主要以植酸〔又稱肌醇六磷酸(C6H18O24P6)〕及其鹽類形式存在,約占有機磷的50%~80%[7]。
植酸分子含有6個磷酸基團和12個可解離質子,具有較強的酸性和金屬螯合能力。因此,植酸的土壤界面過程可影響磷的地球化學行為。因砷與磷是同主族元素,具有某些相似的化學性質和化學行為,故植酸亦可通過界面過程影響砷的賦存形態轉化和生物有效性。土壤/沉積物中植酸主要來源于動植物殘體分解和無機磷的微生物轉化[8]。此外,單胃動物如豬、雞等家禽消化系統中缺乏植酸酶,進食富含植酸的谷物飼料后植酸不能被消化分解,易隨糞便進入土壤,故單胃動物糞便亦是土壤植酸的重要來源,特別是大量、頻繁施用農家肥的農田土壤[9]。因此,植酸磷被認為是農業面源污染的重要來源[10]。已有研究表明,植酸在土壤金屬礦物表面的吸附能力是無機磷的4倍[11]。植酸通過酸化、吸附競爭、置換、溶解金屬等過程影響土壤/沉積物中砷、磷的賦存形態、遷移轉化和生物有效性,引起潛在砷污染和農業面源及水體富營養化污染風險。因此,基于總結分析土壤和沉積物中砷、磷、植酸的含量、來源與賦存形態研究進展,重點闡述植酸對砷、磷賦存形態轉化的影響及機制,以期為合理利用畜禽糞肥及降低砷、磷污染風險提供理論依據和科學支撐。
1 土壤/沉積物中砷和磷的含量、來源與賦存形態
1.1 土壤/沉積物中砷來源、含量與賦存形態
土壤中砷來源復雜,可分為自然來源和人為來源。自然來源主要為母巖,其次為地殼變動、火山爆發、巖石風化、土壤侵蝕等。人為來源主要包括礦產開采(如雄黃、雌黃、砷礦等)、化石燃料燃燒及農業和林業農藥、殺蟲劑、肥料使用,其中農業過程中農藥、化肥使用是農田土壤砷污染的主要來源[12]。
我國表層土壤砷含量均值為3.74~51.9 mg·kg-1,表層土壤砷含量較高省份主要分布于中南和西南部,其中湖南省較高(51.9 mg·kg-1),海南省較低(3.74 mg·kg-1)[13]。此外,礦區周邊土壤砷含量通常較高。例如,云南個舊錫礦區周邊土壤砷含量達162 mg·kg-1,河北半壁山金礦周邊土壤砷超標率達54.9%[14]。
土壤中的砷形態包括價態和賦存形態,亦可分為無機砷和有機砷,無機砷毒性遠高于有機砷。無機砷包括3價砷(As3+)和5價砷(As5+),有機砷主要包括一甲基砷(monomethylarsine,MMA)、二甲基砷(dimethylarsine,DMA)、砷甜菜堿(arsenobetaine,AsB)、砷膽堿(arsenocholine,AsC)和砷糖(arsenosugar,AsS)。As3+的毒性約為As5+的60倍,是甲基砷(一甲基砷和二甲基砷)毒性的70倍[15]。土壤中砷形態受多種因素影響,好氧環境下土壤中砷以As5+為主,厭氧環境下以As3+為主[16]。當土壤pH值為4~8時,砷的常見形態為H3AsO3、H2AsO4、HAsO42-。楊明等[17]指出,微生物對砷的直接還原作用及對固砷礦物的轉化均可引起砷的活化與釋放,同時亦對砷在土壤中的結合形態產生影響,進而影響砷的生物有效性、環境行為及其在不同形態之間的再分配。例如,微生物可將As5+還原為活動性更強的As3+,引起砷的活化與釋放,提高其生物有效性,而土壤中氧化錳可將As3+氧化為As5+,降低其活動性和生物有效性[17]。此外,土壤中陽離子(K+、Na+、Ca2+、Mg2+)越多,砷越易被吸附,其活動性越低。
環境中砷通過各種途徑進入水體后,迅速由水相轉入固相,與懸浮物結合,最終進入沉積物,沉積物是水環境中砷重要的匯[18]。沉積物中砷的活動性、生物有效性和生物毒性不僅與砷總量有關,更取決于其賦存形態。沉積物的pH值、溫度和有機質含量等影響砷的賦存形態轉化[19]。SMEDLEY等[20]研究發現,未受污染湖泊沉積物中砷含量約為0.5~44 mg·kg-1,世界河流沉積物中砷含量均值約為5 mg·kg-1。英格蘭和威爾士河流沉積物中砷含量為5~8 mg·kg-1[21]。DATTA等[22]發現,恒河、布拉馬普特拉河和梅克納河沉積物中砷含量分別為1.2~2.6、1.4~5.9和1.3~5.6 mg·kg-1。我國河流沉積物中砷含量為2.2~285 mg·kg-1,其中陽宗海、滇池、黃浦江、大沙河水系沉積物砷含量較高,分別為54.9~193、7~162、18.7~169 和9.4~285 mg·kg-1(表1)[23]。
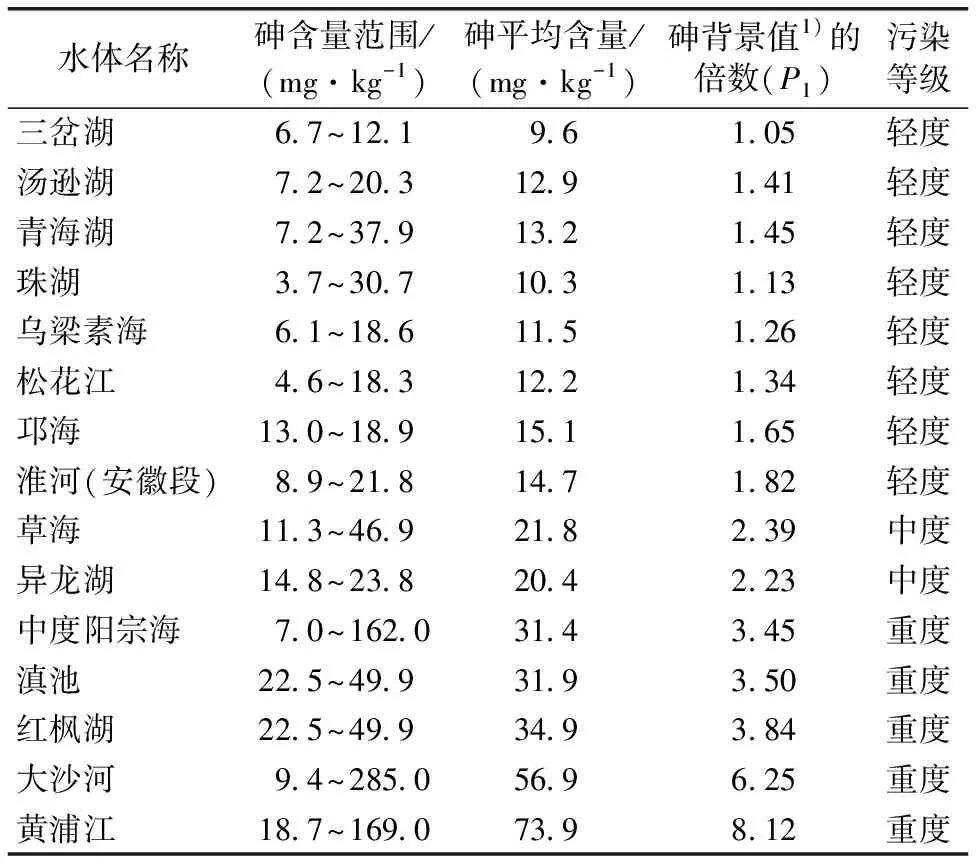
表1 中國部分河流沉積物中砷含量
沉積物中砷通常與鐵-鋁-錳(氫)氧化物、硫化物、無機磷、碳酸鹽、黏土礦物及有機物結合形成不同賦存形態。由于不同研究者對砷形態的理解不同而產生形態分類差異。CUI等[24]根據砷與磷相似的特點,將砷的賦存形態劃分為溶解態砷、弱結合態砷、砷酸鋁、砷酸鐵、砷酸鈣、閉蓄態砷(包裹在鐵氧化物顆粒內部)、有機砷和殘渣態砷。WENZEL等[25]把土壤/沉積物中砷劃分為非專性吸附態砷,專性吸附態砷,與非結晶和結晶水合鐵、鋁氧化物結合態砷,與晶體鐵、鋁氧化物結合態砷和殘渣態砷,其生物有效性逐次降低。
1.2 土壤/沉積物中磷來源、含量與賦存形態
土壤中磷的天然來源主要為巖石風化,其次為降塵、大氣降水、地表徑流、動植物和微生物殘體及施肥、污灌等[26],其中磷肥是主要來源之一。由于土壤中含有大量鐵-鋁-錳水合氧化物和土壤膠體,對磷具有較強的吸附、沉淀作用。磷的各種化合物極易轉變為緩效態或無效態化合物而在土壤中累積,不能被植物直接吸收利用。因此,為了保障農作物產量,農業生產過程中通常施用過量磷肥,而磷肥利用率僅為10%~25%[27],進一步造成磷在土壤中的累積。土壤中大量磷可隨降水進入地表/地下水體,具有潛在面源污染和水體污染風險[28]。
地殼中磷含量約為12 000 mg·kg-1,世界土壤磷含量為200~5 000 mg·kg-1,均值為500 mg·kg-1,我國土壤磷含量約為200~1 100 mg·kg-1。土壤中磷含量取決于母質類型、風化程度和淋失程度。然而,農田土壤磷含量除受成土母質影響外,亦受有機質和磷肥施用的影響。隨著化肥工業的快速發展,我國磷肥用量大幅增加,2018年我國磷肥(P2O5)表觀消費量達1.18×1010kg[29],因磷極易通過吸附和沉淀被固定,導致其作物利用率極低,造成農田土壤磷積累。土壤磷主要分為有機磷和無機磷,無機磷約占土壤總磷的50%~80%,其形態可分為水溶態磷、吸附態磷和礦質態磷[30]。土壤有機磷約占總磷的20%~80%,隨土壤質地等因素變化較大[7,31]。
沉積物作為水體中磷的源與匯,既可接收來自水體沉降和顆粒物中的磷,亦可在特定條件下釋放磷進入水體參與再循環[32]。湖泊沉積物是流域磷循環的重要歸屬,也是湖泊內源性磷的主要來源。通常,湖泊沉積物中磷含量為380~1 210 mg·kg-1,占湖泊總磷的90%[33]。例如,洱海表層沉積物中磷含量為419~1 108 mg·kg-1[34]。沉積物中磷主要可分為無機態和有機態兩大類。無機態磷成分多且種類復雜,主要包括與鈣、鐵、鋁、鎂、錳等元素共沉淀而形成的礦物態磷,沉積物表面吸附的可交換態磷以及沉積物間隙水中的溶解態磷。有機態磷主要包括肌醇磷酸、磷脂類、核酸類、磷蛋白、微生物磷和卵磷脂等磷脂化合物[35]。
沉積物中磷形態的主要影響因素包括沉積物母質來源、pH值、氧化還原電位(Eh)、溫度、有機質含量、陽離子交換容量、粒徑組成等[36]。pH值主要通過影響離子交換過程進而影響Fe、Al、Ca與磷的結合狀態。在pH中性環境中,磷主要以HPO42-、H2PO4-形態存在,極易被土壤/沉積物固相吸附,升高(>7.14)或降低pH值(<6.99)均可促進固相磷的釋放[37]。pH值>7時,磷釋放以離子交換為主,促進OH-與Fe、Al結合態磷的交換,引起磷釋放[38-39];pH值<6.99時,磷釋放以溶解為主,促進Ca結合態磷(Ca-P)釋放[38]。堿性(pH值為7.2~11.8)條件下,沉積物中Fe-P釋放明顯,導致上覆水中磷濃度升高,堿性越強,磷釋放量越大[40]。當pH值為4~8時,沉積物磷釋放量受pH值影響較小(0.53 mg·kg-1);而當pH值≥8時,磷釋放量明顯提高至9.97 mg·kg-1。
2 土壤/沉積物中植酸含量與賦存形態
2.1 土壤中植酸來源、含量與賦存形態
土壤中有機磷主要以植酸形式存在,通過脫磷酸化過程逐次釋放磷,土壤中肌醇一磷酸到肌醇五磷酸(IP1-5)含量較少[41]。如圖1所示,土壤中植酸具有4種異構體,其中以myo異構體為主,占比為56%~90%;其次為scyllo、D-chiro、neo異構體,占比分別為20%~50%、6%~10%和1%~5%。土壤中植酸磷年積累量達5.1×107t,相當于磷肥施用量的65%[42]。土壤中植酸主要來自植物、單胃動物糞便及微生物[8]。作為種子萌發的磷儲備庫,谷物和種子中含有大量植酸鹽,約占總磷的80%和有機磷的90%~100%[43]。土壤中植酸磷約占有機磷的50%和總磷的80%,因此是植物磷營養的潛在重要來源[44]。單胃動物(家禽、豬、牛、羊等)體內缺少植酸酶,不能有效分解利用谷物飼料中的植酸,導致大量植酸隨糞便進入土壤[45],是農田土壤中植酸輸入的重要來源。此外,L′ANNUNZIATA等[46]發現,隱球菌(CryptococcusMelibiosum)可利用土壤中14C標記的myo-植酸差向異構化合成D-chiro-植酸,表明微生物對植酸的合成和同分異構體轉化具有一定作用。
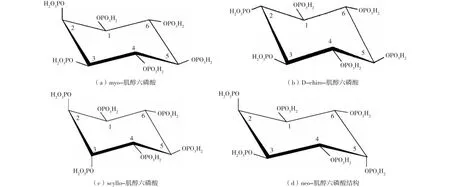
圖1 myo-肌醇六磷酸、D-chiro-肌醇六磷酸、scyllo-肌醇六磷酸和neo-肌醇六磷酸結構[42]
土壤中植酸含量因土地利用類型、土壤性質及提取分析方法而異(表2)。例如,耕地土壤中植酸含量為17~142 mg·kg-1,作物和牧場土壤中為42~220 mg·kg-1。單胃動物糞便中植酸含量高達153~1 325 mg·kg-1,豬、牛和家禽糞便中植酸磷含量均值分別為457、1 047和2 277 mg·kg-1,占總磷的16%~17%。澳大利亞牧場土壤水提取物中植酸酶可水解磷含量為0.1~0.4 mg·kg-1,而智利南部牧場牛糞的NaOH-EDTA提取物中植酸酶可水解磷含量為153~613 mg·kg-1,占總磷含量的9%~14%、有機磷含量的44%~73%[47]。TURNER[48]發現,英格蘭和威爾士溫帶牧場土壤中植酸含量達26~189 mg·kg-1,占有機磷總量的11%~35%,且植酸含量與草酸鹽提取的鐵鋁含量呈正相關,與碳/有機磷含量之比呈負相關,與總碳、總氮、黏土礦物含量及微生物量無相關性。

表2 不同類型土壤中植酸含量及總有機磷占比[48,53-58]
2.2 沉積物中植酸來源、含量與賦存形態
土壤中的大量植酸可隨地表、地下水進入河流湖泊沉積物[49]。此外,沉積物中植酸亦來自湖泊內藻類等大型植物及污水排放、工業和動物廢棄物(直接排泄物、泥漿)。沉積物中植酸主要為鈣、鎂、鐵、錳等難溶性或不溶性金屬絡合物[50],其含量因地區來源、提取和分析方法而異(表3)。例如,DE GROOT等[51]分析法國卡瑪格湖沉積物中植酸含量為24~149 mg·kg-1,占總磷的17.6%;美國門多塔湖沉積物中植酸含量為38~51 mg·kg-1,占總磷的9.3~12.7%[50]。在東京灣淺海沉積物中發現植酸的3種同分異構體(myo、scyllo、chiro),該3種異構體主要來自于陸生植物而非浮游動植物,在流向海灣的主要河流流域的表層沉積物中植酸含量為7.8~175 mg·kg-1,而海灣表層(0~0.25 cm)沉積物中為0.31~3.1 mg·kg-1,占有機磷的0.23%[49,52]。植酸在海洋環境中易受微生物降解的影響,故含量較低[52]。
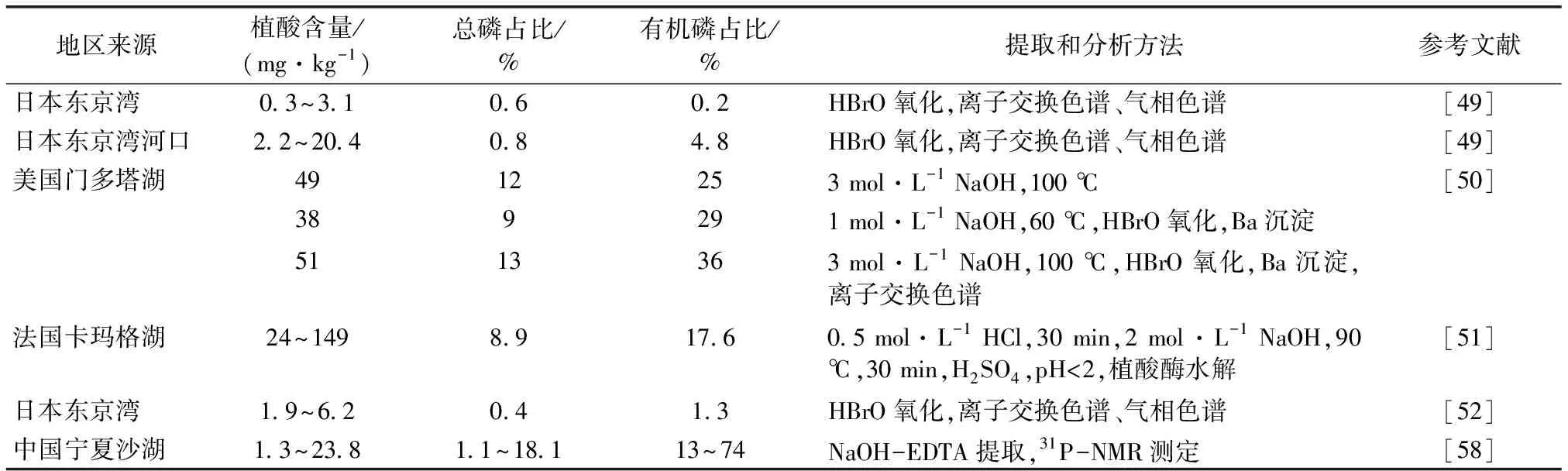
表3 不同地區沉積物中植酸含量及總磷占比[49-52,58]
SUZUMURA等[52]研究發現,東京灣表層沉積物中植酸含量較低(2.48 mg·kg-1),且隨深度增加而降低。COSGROVE[59]認為,酸性土壤中主要為植酸鐵/鋁,在中性和堿性土壤中主要為植酸鈣,在厭氧和堿性條件下植酸鈣可發生礦化。沉積物通常為缺氧條件,海水pH值一般為堿性,因此,海洋環境利于植酸的礦化反應。東京灣低植酸含量可歸因于生物降解,由陸地輸送至海岸沉積物的植酸在表層含氧層內礦化,在底層缺氧區厭氧分解過程中完全礦化,過程中釋放大量無機磷進入上覆海水中,或重新分配形成沉積物中的次生礦物(磷酸鐵和氟磷礦等)[52,60]。
3 植酸對土壤/沉積物中砷、磷生物有效性的影響與機制
3.1 植酸對砷賦存形態轉化和生物有效性的影響與機制
有機酸可通過酸化、絡合、溶解等過程影響重(類)金屬的溶解性和活動性[60]。鐵/鋁氧化物對砷的吸附是控制其在水相/固相分配的重要過程[58]。不同有機酸對土壤中鐵/鋁結合砷的溶解釋放能力從大到小表現為植酸(9.8%)>檸檬酸(7.5%)>蘋果酸(1.3%)>乳酸(0.9%)>乙酸(0.5%)[60]。
砷的賦存形態和穩定性受土壤/沉積物pH值和競爭性陰離子的影響,因砷與磷為同主族類似物,無機磷對砷的影響已有大量研究,而有機磷(特別是植酸)對砷賦存形態和有效性的影響研究較少。植酸結構中的6個磷酸基團和12個可解離質子使其具有多陰離子性質,易被土壤固相吸附和與金屬陽離子絡合,酸性條件下主要為Fe/Al絡合物,堿性條件下主要為Ca/Mg絡合物[61]。植酸與砷在鐵(氫)氧化物表面發生吸附競爭,因植酸與Fe的復合物穩定性更強,植酸對難溶性FeAsO4的溶解率高達39%[62]。
BALINT等[63]研究植酸對As3+和As5+在針鐵礦上吸附和固定的影響,發現植酸可有效置換和抑制砷在針鐵礦表面的吸附,而As3+和As5+對植酸進行置換或抑制吸附的影響較小。植酸對針鐵礦吸附As3+和As5+的置換和抑制作用受pH值、停留時間和陰離子添加順序的影響[64]。隨pH值升高,植酸對As3+的解吸率降低,對As5+的解吸率提高。例如,當溶液pH值由4.5升至8.5,針鐵礦對As3+的吸附量由2.4提高至3 μmol·m-2,對As5+的吸附量由2.5降至1.4 μmol ·m-2,可歸因于針鐵礦-As配合物解離常數的差異(As3+:pKa1=9.2,pKa2=12.7,pKa3=13.4;As5+:pKa1=2.3,pKa2=6.8,pKa3=11.8)[65],針鐵礦對As3+的結合強度隨pH值升高而增強。此外,針鐵礦吸附As3+后通常帶正電荷,而吸附As5+后帶負電荷。帶正電荷的針鐵礦-As3+配合物易與帶負電荷的植酸結合,植酸可在酸性條件下通過置換解吸93%的As3+。針鐵礦-As5+配合物帶負電荷,植酸對As5+的置換解吸率隨pH值升高而升高,可能是由于針鐵礦的ζ電位隨pH值升高而降低,降低了對As5+的吸附作用。故隨pH值升高,植酸對As5+的解吸率提高,該結果與ZANZO等[66]研究一致,即植酸在酸性和堿性條件下易從針鐵礦-As配合物中分別置換解吸As3+和As5+。停留時間對植酸從礦物表面置換解吸As3+和As5+的影響表現為隨停留時間的增加而降低。PIGNA等[67]發現,停留時間為1 h時,植酸對針鐵礦表面As3+和As5+的解吸率分別為87%和61%,但停留時間為30 d時,植酸對針鐵礦吸附態As無顯著影響。此外,陰離子添加順序亦可影響植酸對礦物表面As的解吸效率,在針鐵礦-As配合物體系中加入植酸、植酸-針鐵礦體系中加入As3+/As5+、針鐵礦體系中同時加入植酸和As3+/As5+情況下,針鐵礦對As3+吸附量均高于As5+,進一步表明As3+可利用不同于植酸的吸附位點,而As5+與植酸競爭同一吸附位點。CELI等[64]發現,植酸通過與鐵絡合可導致鐵氧化物聚集分散和溶解,從而釋放鐵砷共沉淀中的As。
綜上,土壤和沉積物中植酸對砷賦存形態轉化與生物有效性的影響機理主要包括:(1)植酸與As3+或As5+競爭礦物表面吸附位點,通過置換、抑制吸附進而提高As的活動性;(2)植酸與鐵、鋁氧化物及水化物形成穩定螯合物,通過改變礦物表面電荷影響其對As的吸附;(3)植酸的酸化作用使土壤pH值降低,酸性條件下促進As3+的解吸釋放;(4)植酸通過促進鐵鋁氧化物溶解,進而促進鐵/鋁-砷共沉淀中As的釋放(圖2)。
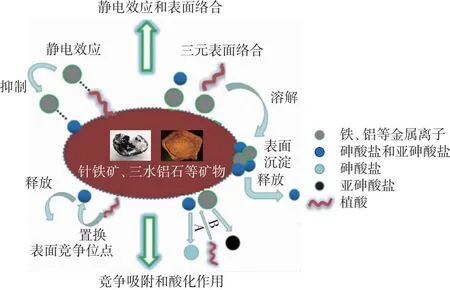
A—酸性條件下,植酸促進As5+釋放;B—堿性條件下,植酸促進As3+釋放。
3.2 植酸對磷賦存形態轉化和生物有效性的影響與機制
與無機磷在土壤礦物上的吸附機制類似,植酸可通過配體交換在鐵鋁氧化物礦物表面吸附,形成多元表面配合物。植酸通過競爭吸附位點誘導磷解吸,提高孔隙水中無機磷的含量[68]。因含有6個磷酸基團,植酸更易在礦物表面發生吸附。例如,方解石對植酸磷的吸附量(17.8 μmol·m-2)是無機磷(1.4 μmol·m-2)的12.7倍。此外,金屬氫氧化物比硅酸鹽對植酸的吸附性更強。例如,植酸在非晶態氫氧化鋁表面的最大吸附量(51.5 μmol·m-2)顯著高于高嶺石(0.22 μmol·m-2)[69]。植酸影響無機磷在礦物表面吸附的因素主要包括:(1)植酸含量;(2)無機磷與植酸的添加順序;(3)環境pH值;(4)平衡時間(圖3)。因植酸比無機磷的親和力更強,無定形氫氧化鋁對無機磷的吸附量隨植酸含量升高而降低[70-71]。

A—初始植酸/磷比值較高,先加入植酸,平衡時間較長;B—初始植酸/磷比值較低,先加入磷,平衡時間較短。
此外,pH值對無機磷的影響比植酸更顯著,當pH值由4.0提高至10.0時,氫氧化鋁對植酸和無機磷的吸附量分別降低53.5%和81.4%。在添加順序上,GUAN等[70]發現,先添加植酸后加入無機磷對吸附態植酸無顯著影響,而在植酸之前或與植酸一起加入無機磷,植酸在氫氧化鋁上的吸附量下降約50%,表明無機磷難以取代已吸附的植酸。ANDERSON[71]等研究平衡時間對土壤吸附植酸和無機磷的影響,發現其吸附平衡時間分別為72和24 h。KARATHANASIS等[72]研究植酸和無機磷在土壤礦物(蛭石、蒙脫石、三水鋁石和針鐵礦)的競爭吸附,發現較長的平衡時間顯著提高了植酸在礦物表面的吸附量。兩項研究均表明,植酸的吸附平衡時間較長,與無機磷相比,植酸具有更大電荷密度和空間位阻,故增加了其達到平衡所需的時間。因此植酸比無機磷具有更強的吸附能力,可解吸礦物表面吸附態無機磷[73]。
3.2.1鐵鋁氧化物表面競爭吸附
酸性土壤對植酸的吸附作用主要依賴于鐵鋁氧化物,尤其是弱晶質鐵鋁氧化物[72]。與針鐵礦和赤鐵礦等晶質氧化鐵相比,弱晶態和無定形氧化鐵(如水鐵礦)對植酸的吸附能力更強。由K值可知,鐵鋁氧化物對植酸的親和力顯著高于無機磷(表4)。由于弱晶質鋁氧化物活性位點密度更大,其對植酸的吸附量顯著高于剛玉和勃姆石。YAN等[74]發現,無定形氫氧化鋁對植酸的吸附量(16 μmol·m-2)顯著高于剛玉(1.13 μmol·m-2)和勃姆石(0.73 μmol·m-2)。
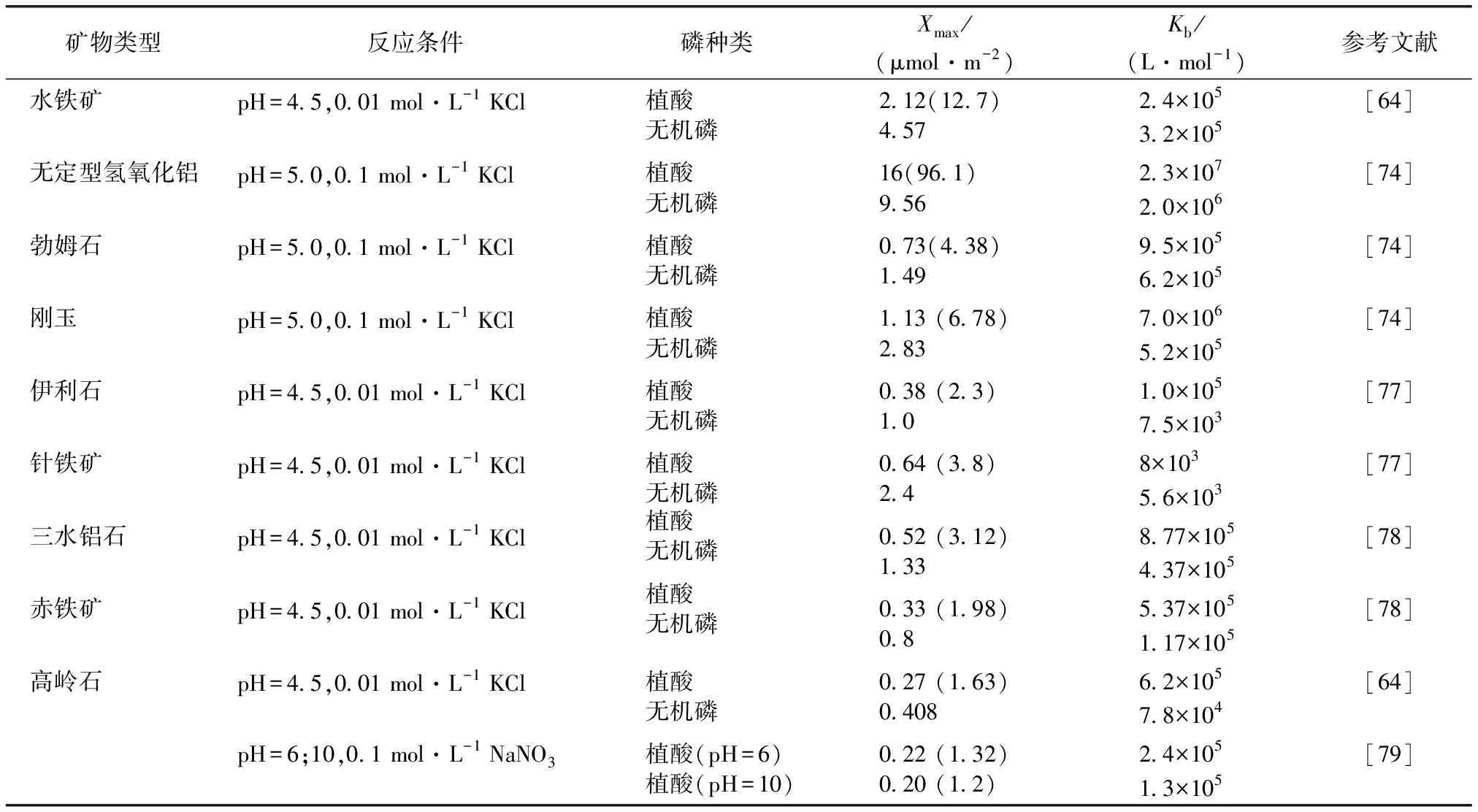
表4 植酸和無機磷的朗繆爾等溫吸附參數[64,77-79]
pH值、電解質及共存離子影響植酸和磷在鐵鋁氧化物的表面吸附。CELI等[75]發現,電解質為KCl時,隨pH值升高吸附量降低,針鐵礦對植酸的吸附量由4.5降至0.18 μmol·m-2,對無機磷的吸附量由2.5降至0.67 μmol·m-2,對植酸的影響更為顯著。而電解質為CaCl2時,隨pH值升高,針鐵礦對植酸的吸附量由4.5提高至4.9 μmol·m-2,無機磷吸附量由2.7提高至4.8 μmol·m-2。鐵鋁氧化物通過表面H2O和—OH與磷酸基配位交換來吸附植酸和磷[76]。植酸在鐵鋁氧化物表面的吸附包括表面絡合和沉淀反應2個過程。首先,植酸通過配體交換在鐵鋁氧化物表面形成單齒或多齒絡合物,吸附態植酸為游離態金屬離子提供吸附位點,進而形成三元配合物,通過聚合或縮合轉化為表面析出物,形成植酸鹽沉淀。
3.2.2中性和堿性土壤競爭吸附
在中性至堿性土壤中鐵鋁氧化物含量降低。在堿性土壤中磷通常與鈣等堿土金屬絡合[80]。此時,植酸的吸附主要受黏土礦物和有機質含量的影響[74,81]。MCKERCHER等[81]研究表明,中性和堿性土壤對植酸具有較高的吸附能力。PRIETZE等[82]發現,中性土壤中有機結合態鈣主要以植酸鈣形式存在。在鈣質土壤中,方解石是吸附磷的主要成分,反應過程包括低濃度磷吸附和高濃度磷酸鈣沉淀。
無機磷在方解石表面的作用過程已有較多研究,但目前對有機磷(植酸)的研究較少,已有研究主要關注不同類型土壤和方解石對植酸的吸附特性。例如,CELI等[83]發現,方解石對植酸的吸附量顯著高于無機磷,吸附平衡時植酸和無機磷吸附密度分別為17.8和1.4 μmol·m-2,但當平衡濃度大于6×10-4mol·L-1時,由于形成磷酸鈣沉淀,無機磷吸附密度急劇升高,達到155 μmol·m-2。方解石對植酸的吸附固定能力導致其在土壤中積累并影響磷的生物有效性,同時引起礦物表面電化學性質、顆粒大小以及分散穩定性的變化。CHEN等[84]研究發現,不同于鐵鋁氫氧化物的內球絡合形式,植酸主要通過外球絡合在方解石表面發生吸附。WAN等[85]發現,在pH值為8.5條件下,方解石對植酸的吸附速率顯著高于無機磷,植酸與方解石相互作用形成球形沉淀物,伴隨方解石的溶解和Ca2+的再沉淀,無機磷被釋放。
4 結語與展望
植酸隨動植物殘體分解、微生物轉化及禽畜糞便等途徑進入到土壤和沉積物中,易在礦物表面吸附或與金屬離子形成難溶性絡合物,通過界面反應影響砷和磷的賦存形態和生物有效性,引起潛在砷污染和農業面源及水體富營養化污染風險。因此,該文通過闡述土壤和沉積物中砷、磷、植酸的含量、來源與賦存形態,重點綜述植酸對砷、磷賦存形態轉化的影響因素(溫度、植酸濃度、環境pH值、平衡時間、離子添加順序等)及機制(酸化、吸附競爭、置換、溶解等),對有效阻控土壤和水體砷/磷污染具有重要的現實意義。
以下方面可進一步深入研究:
(1)應采用先進表征技術,如衰減全反射傅里葉變換紅外光(ATR-FTIR)、核磁共振(NMR)和X-射線吸收光譜(XAS)等,結合量子化學理論計算和電荷分布-多位點絡合模型或三層模型,從分子水平深入系統地揭示植酸的固液界面反應特性與機制。應用光譜技術可表征植酸和砷、磷在不同界面上反應的絡合形態,結合表面絡合模型進一步分析界面上不同形態砷、磷的電荷分布、反應常數、吸附量或吸附比例,解釋宏觀吸附行為和吸附機制。
(2)植酸的強絡合能力提高了重金屬在土壤界面的遷移性,對重金屬污染土壤中金屬的提取去除具有積極意義,可用于污染場地和土壌的修復,但是關于植酸與砷共存時礦物對砷吸附解吸行為的研究還比較缺乏。應從多角度揭示植酸對土壤砷的活化效果,如與微生物生理生態結合,進行根際微生物的代謝通路、蛋白及酶分泌、基因表達等方面的研究,從微觀層面上進一步探究植酸對砷的活化機理。
(3)開展廢棄物來源植酸的相關研究。植酸的來源不再局限于單純的外源添加,而是充分利用可以產生植酸的農林廢棄物,如禽畜糞便、污泥等,研究其活化砷、磷的效果并與單純施加植酸進行比較,有助于拓寬不同來源植酸的研究,形成實用技術。
