抑制泛素特異性蛋白酶7改善血管緊張素Ⅱ誘導(dǎo)的心肌細(xì)胞肥大
2024-04-29 16:00:05陳夢(mèng)雅謝賽陽(yáng)鄧偉
心血管病學(xué)進(jìn)展 2024年2期
關(guān)鍵詞:氧化應(yīng)激
陳夢(mèng)雅 謝賽陽(yáng) 鄧偉

【摘要】目的?探究泛素特異性蛋白酶7(USP7)抑制劑FT671在血管緊張素Ⅱ(AngⅡ)誘導(dǎo)的H9C2細(xì)胞肥大中的作用和潛在機(jī)制。方法?將H9C2細(xì)胞隨機(jī)分為四組:對(duì)照組、FT671組、AngⅡ組、FT671+AngⅡ組。利用α-actinin細(xì)胞免疫熒光染色檢測(cè)心肌細(xì)胞表面積;western blot檢測(cè)心房鈉尿肽(ANP)、腦鈉肽(BNP),B細(xì)胞淋巴瘤2(BCL-2)、B細(xì)胞淋巴瘤2相關(guān)蛋白X(Bax)、白細(xì)胞介素-6(IL-6)、單核細(xì)胞趨化蛋白-1(MCP-1)、腫瘤壞死因子-α(TNF-α)、核苷酸結(jié)合寡聚化結(jié)構(gòu)域樣受體蛋白3(NLRP3)以及NADPH氧化酶4(Nox4)的蛋白表達(dá)水平;RT-PCR檢測(cè) ANP、BNP、肌球蛋白重鏈(β-MHC)、IL-6、MCP-1、TNF-α的mRNA表達(dá);TUNEL染色檢測(cè)細(xì)胞凋亡情況,細(xì)胞計(jì)數(shù)試劑盒8(CCK8)實(shí)驗(yàn)檢測(cè)各組細(xì)胞活力,活性氧(ROS)染色檢測(cè)細(xì)胞內(nèi)氧化損傷水平。結(jié)果?與對(duì)照組相比,AngⅡ組USP7蛋白表達(dá)明顯增加,而使用FT671后,USP7表達(dá)明顯被抑制。與AngⅡ組相比,F(xiàn)T671+AngⅡ組心肌細(xì)胞面積減小;ANP、BNP、Bax的蛋白表達(dá)減少,ANP、BNP、β-MHC、Bax、IL-6、MCP-1以及TNF-α的mRNA表達(dá)減少;BCL-2的蛋白和mRNA表達(dá)均增加。與AngⅡ組相比,F(xiàn)T671+AngⅡ組TUNEL陽(yáng)性細(xì)胞明顯減少,心肌細(xì)胞活力增強(qiáng), ROS生成減少,Nox4、NLRP3蛋白表達(dá)減少,差異具有統(tǒng)計(jì)學(xué)意義(P<0.05)。結(jié)論?FT671通過(guò)抑制Nox4/ROS/NLRP3炎性通路減輕AngⅡ誘導(dǎo)的心肌細(xì)胞中的氧化應(yīng)激和炎性反應(yīng),從而減輕心肌細(xì)胞肥大。
【關(guān)鍵詞】泛素特異性蛋白酶7;血管緊張素Ⅱ;FT671;心肌肥厚;氧化應(yīng)激
【DOI】10.16806/j.cnki.issn.1004-3934.2024.02.000
Inhibition of Ubiquitin-Specific Protease 7 Improves AngiotensinⅡ-Induced Cardiomyocyte Hypertrophy
CHEN Mengya,XIE Saiyang,DENG Wei
(Department of Cardiology,Renmin Hospital?of Wuhan University,Hubei Key Laboratory of Metabolic and Chronic Diseases,Wuhan?430060,Hubei,China)
【Abstract】Objective?To investigate the effect and mechanism of ubiquitin-specific protease 7(USP7) inhibitor FT671 in cardiomyocyte hypertrophy induced by angiotensinⅡ(AngⅡ). Methods?H9C2?cardiomyocytes were randomly divided into four groups:?control group,F(xiàn)T671 group,AngⅡ?group,F(xiàn)T671+AngⅡ?group. The surface area of cardiomyocytes?was detected by immunofluorescence staining of α-actinin. The protein expression of?atrial natriuretic peptide (ANP),brain natriuretic peptide (BNP),B-cell lymphoma-2 (BCL-2),B-cell lymphoma-2-associated X protein (Bax),interleukin-6 (IL-6),monocyte chemoattractant protein-1 (MCP-1),tumor necrosis factor-α (TNF-α),nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) and reduced nicotinamide adenine dinucleotide phosphate oxidase?4?(Nox4) were detected by western blot. The mRNA levels of ANP,BNP,β-myosin heavy chain (β-MHC),IL-6,MCP-1,TNF-α?were detected by Real-Time PCR. TUNEL staining was used to evaluate cell apoptosis. Cell Counting Kit-8(CCK8)?was used to detect cell viability. The fluorescent probe 2',7'-dichlorofluorescein diacetate (DCFH-DA) was used to measure intracellular reactive oxygen species (ROS) formation. Results There was a significant increase in USP7 protein expression in the AngⅡ?group,which was evidently inhibited by the USP7 inhibitor FT671. Compared with AngⅡ?group,cardiomyocyte?surface area was significantly?reduced in FT671+AngⅡ group; the protein expression of ANP,BNP and?Bax,as well as?mRNA level of ANP,BNP,β-MHC,Bax,IL-6,MCP-1,TNF-α,were decreased?in FT671+AngⅡ?group. In addition,the protein expression and mRNA level of BCL-2 were increased in FT671+AngⅡ group. Additionally,compared with AngⅡ?group,TUNEL positive cells was significantly reduced,cell viability was improved and ROS generation was inhibited in FT671+AngⅡ group. Further studies showed the protein expression of NLRP3 and Nox4?was decreased after FT671 treatment in AngⅡ?induced cardiomyocyte hypertrophy.?Conclusion The USP7 inhibitor FT671 attenuated oxidative stress and inflammatory response in AngⅡ-induced cardiomyocyte by inhibiting the Nox4/ROS/NLRP3 inflammatory pathway,thereby reducing cardiomyocyte hypertrophy.
【Keywords】Ubiquitin-specific protease 7;AngiotensinⅡ;FT671;Hypertrophy;Oxidative stress
心力衰竭是各種心臟疾病發(fā)展的終末階段,以心肌肥厚和纖維化為特征的心肌重構(gòu)已被確定為心力衰竭病程進(jìn)展中的決定性因素[1]。生理性心肌肥厚在刺激消失后可以逆轉(zhuǎn),而心肌梗死、高血壓等長(zhǎng)期慢性刺激導(dǎo)致的病理性心肌肥厚往往會(huì)導(dǎo)致心肌重構(gòu),產(chǎn)生不良后果,如心律失常、心力衰竭等[2]。心肌肥厚的發(fā)生發(fā)展機(jī)制比較復(fù)雜,至今尚未完全闡明。因此,探討心肌肥厚的發(fā)生發(fā)展機(jī)制,從而尋找更有效的作用靶點(diǎn),抑制甚至逆轉(zhuǎn)心肌重構(gòu)的發(fā)生是針對(duì)心力衰竭的重要策略。
泛素-蛋白酶體系統(tǒng)是細(xì)胞內(nèi)蛋白質(zhì)降解的主要途徑,通過(guò)對(duì)底物蛋白的泛素化降解,參與包括細(xì)胞周期調(diào)節(jié)、免疫反應(yīng)、細(xì)胞受體功能及腫瘤生長(zhǎng)等在內(nèi)的多種細(xì)胞活動(dòng)[3]。去泛素化酶(deubiquitinating enzyme,DUB)家族可將泛素分子從泛素化標(biāo)記的蛋白質(zhì)或者前體蛋白上水解下來(lái),起到去泛素化的作用,對(duì)蛋白降解進(jìn)行反向調(diào)節(jié)[4-5]。既往研究[6-8]顯示,DUB在心肌肥厚、心肌梗死、心房顫動(dòng)等心血管疾病中起著至關(guān)重要的作用。泛素特異性蛋白酶7(ubiquitin-specific protease 7,USP7)是第一個(gè)被發(fā)現(xiàn)的DUB,主要位于細(xì)胞核中,參與細(xì)胞增殖、DNA損傷反應(yīng)、腫瘤發(fā)生、炎癥和細(xì)胞凋亡等過(guò)程[9-10],在神經(jīng)退行性疾病和癌癥中被廣泛研究,USP7的抑制劑也常作為抗腫瘤藥物被廣泛研究[11-12],但是在心血管系統(tǒng)疾病中研究較少。最近研究[13]發(fā)現(xiàn)USP7在心肌細(xì)胞缺氧條件下的表達(dá)顯著增加,可能參與心肌梗死和心肌缺血再灌注損傷過(guò)程。然而,關(guān)于USP7在心肌肥厚中的作用和分子機(jī)制知之甚少。在本研究中,應(yīng)用USP7特異性抑制劑FT671干預(yù)血管緊張素Ⅱ(angiotensinⅡ,AngⅡ)誘導(dǎo)的H9C2心肌細(xì)胞肥大,探討USP7在心肌細(xì)胞病理性肥大中的作用和作用機(jī)制。
1??材料和方法
1.1??實(shí)驗(yàn)材料
試劑:H9C2心肌細(xì)胞購(gòu)自中國(guó)科學(xué)院細(xì)胞庫(kù)(上海,中國(guó))。FT671(HY-107985)?購(gòu)自美國(guó)MedChemexpress(MCE)公司,AngⅡ(美國(guó)Sig-maAldrich公司),胎牛血清和F12培養(yǎng)液(美國(guó)Gibco公司),USP7抗體(ABclonal,A3448),α-actinin、心房鈉尿肽(ANP)、腦尿鈉肽(BNP),B細(xì)胞淋巴瘤2(BCL-2)、B細(xì)胞淋巴瘤2相關(guān)蛋白X(Bax)抗體(美國(guó)abcam公司),核苷酸結(jié)合寡聚化結(jié)構(gòu)域樣受體蛋白3(NLRP3),羊抗兔二抗(美國(guó)Cell Signaling Technology公司),NADPH氧化酶4(Nox4)(Proteintech)。TUNEL染色試劑盒,細(xì)胞計(jì)數(shù)試劑盒8(CCK8)試劑盒,活性氧(reactive oxygen species,ROS)檢測(cè)試劑盒(Beyotime Biotechnology)。Trizol(Invitrogen),反轉(zhuǎn)錄試劑盒(瑞士Roche公司),Radioimmunoprecipitation Assay Buffer(RIPA)裂解液(美國(guó)賽默飛世爾公司)。
儀器:凝膠電泳系統(tǒng)(北京六一儀器廠),凝膠電泳轉(zhuǎn)移系統(tǒng)(BIO-RAD),聚偏二氟乙烯膜(美國(guó)Milipore),化學(xué)發(fā)光蛋白檢測(cè)顯影儀(美國(guó)Borad),熒光顯微鏡(OLYMPUS DX51),SYBRGreen I Master(瑞士Roche公司,4707516001),LightCycler480 SYBRGreen熒光定量?jī)x(瑞士Roche公司,型號(hào):LC480),酶標(biāo)儀(美國(guó)伯騰儀器有限公司,Synergy HT)。
1.2??方法
1.2.1??細(xì)胞培養(yǎng)與分組
使用含10%胎牛血清的DMEM培養(yǎng)基,于37?°C、95%O2、5%CO2培養(yǎng)箱中培養(yǎng)H9C2心肌細(xì)胞,待細(xì)胞貼壁之后長(zhǎng)至90%密度左右,用0.25% 胰蛋白酶消化傳代,待細(xì)胞長(zhǎng)至培養(yǎng)皿面積的70%~80%時(shí),進(jìn)行以下實(shí)驗(yàn):
將H9C2心肌細(xì)胞隨機(jī)分為4組(n=6):空白對(duì)照組,F(xiàn)T671(10 μmol/L)組,AngⅡ(1 μmol/L)組,F(xiàn)T671(10 μmol/L)+?AngⅡ(1 μmol/L)組。待細(xì)胞長(zhǎng)至培養(yǎng)皿面積的70%~80%,換液之后根據(jù)分組加入生理鹽水或AngⅡ和/或FT671,培養(yǎng)24?h之后收集細(xì)胞進(jìn)行后續(xù)實(shí)驗(yàn)。
1.2.2??細(xì)胞免疫熒光檢測(cè)細(xì)胞面積
采用細(xì)胞爬片,用4%多聚甲醛固定細(xì)胞,0.2%Triton-X-100通透細(xì)胞,10% 羊血清室溫孵育60?min進(jìn)行封閉。α-actinin一抗(1﹕200)4?°C 孵育過(guò)夜,綠色熒光標(biāo)記的二抗(1﹕100)濕盒中37?°C 孵育60?min。滴加封片劑封片,放置10?min左右后,在熒光顯微鏡下觀察、拍照,并使用Image J軟件分析,計(jì)算各組心肌細(xì)胞肥大面積。
1.2.3??免疫印跡法western blot檢測(cè)目的蛋白表達(dá)
提取各組心肌細(xì)胞蛋白,采用BCA法進(jìn)行定量,制備上樣蛋白,使用10% 十二烷基硫酸鈉-聚丙烯酰胺(SDS-PAGE)電泳分離蛋白,轉(zhuǎn)膜至聚偏氟乙烯(PVDF)膜,5%脫脂牛奶封閉1?h后,孵育一抗置于4?°C過(guò)夜。用Tris緩沖液加Tween 20液洗膜3次后,二抗室溫孵育1?h,使用化學(xué)發(fā)光后成像系統(tǒng)進(jìn)行圖像采集,Image J軟件對(duì)蛋白表達(dá)水平進(jìn)行定量,β-tubulin作為內(nèi)參蛋白。
1.2.4??實(shí)時(shí)定量PT-PCR法檢測(cè)目的基因mRNA表達(dá)
按照 Trizol (Invitrogen)法提取細(xì)胞總mRNA,紫外分光光度計(jì)檢測(cè)RNA含量及純度。每個(gè)樣本取2?μg總mRNA,使用Roche反轉(zhuǎn)錄試劑盒(20?μL反應(yīng)體系)反轉(zhuǎn)錄,之后進(jìn)行PCR擴(kuò)增,以GAPDH作為內(nèi)參基因,計(jì)算各組相較于對(duì)照組的相對(duì)表達(dá)量,計(jì)算公式為2-ΔΔCt(表1)。
1.2.5??TUNEL染色檢測(cè)心肌細(xì)胞凋亡
處理后的細(xì)胞用4%多聚甲醛固定細(xì)胞30?min,0.2%Triton-X-100通透細(xì)胞,每組加50?μL TUNEL檢測(cè)液,37?℃避光孵育60?min。滴加含有DAPI的封片劑封片,放置10?min左右后,在熒光顯微鏡下觀察、拍照,并使用Image?J軟件分析,計(jì)算各組每個(gè)視野TUNEL陽(yáng)性細(xì)胞占總細(xì)胞的比率即凋亡率。
1.2.6??CCK8檢測(cè)心肌細(xì)胞活性
使用CCK8試劑盒檢測(cè)細(xì)胞活性。于96 孔板中每孔加入100?μL(約4?000個(gè)細(xì)胞)H9C2細(xì)胞懸液,置于37?℃、5% CO2培養(yǎng)箱中培養(yǎng)12?h,待細(xì)胞貼壁后根據(jù)分組進(jìn)行處理,培養(yǎng)24?h后每孔加入10?μL CCK8試劑,繼續(xù)置于培養(yǎng)箱內(nèi)孵育2?h。用酶標(biāo)儀檢測(cè),分別于450?nm波長(zhǎng)處測(cè)定光密度(optical density,OD)值,計(jì)算細(xì)胞活性。實(shí)驗(yàn)結(jié)果以細(xì)胞存活率表示,細(xì)胞存活率=(實(shí)驗(yàn)組OD值-空白對(duì)照組OD 值)/(對(duì)照組OD 值-空白對(duì)照組OD值)× 100%。
1.2.7??ROS實(shí)驗(yàn)檢測(cè)氧化損傷
使用熒光探針DCFH-DA測(cè)量細(xì)胞內(nèi)ROS的形成。用10 μmol/L DCFH-DA處理細(xì)胞爬片并置于37?℃細(xì)胞培養(yǎng)箱內(nèi)孵育20?min,用無(wú)血清培養(yǎng)液洗滌三次。用倒置顯微鏡記錄心肌細(xì)胞中ROS的熒光,并用Image J軟件進(jìn)行分析。
1.3??統(tǒng)計(jì)學(xué)處理
應(yīng)用Graph Pad Prism 9.0統(tǒng)計(jì)學(xué)軟件對(duì)數(shù)據(jù)進(jìn)行統(tǒng)計(jì)分析,Image J軟件分析條帶灰度值,Adobe Illustrator軟件進(jìn)行圖形繪制。數(shù)據(jù)采用均數(shù)±標(biāo)準(zhǔn)差(![]() ±s)表示,多組比較采用單因素方差分析(one-way ANOVA),兩組間比較采用成對(duì)t檢驗(yàn),以P<0.05為差異有統(tǒng)計(jì)學(xué)意義。
±s)表示,多組比較采用單因素方差分析(one-way ANOVA),兩組間比較采用成對(duì)t檢驗(yàn),以P<0.05為差異有統(tǒng)計(jì)學(xué)意義。
2??結(jié)果
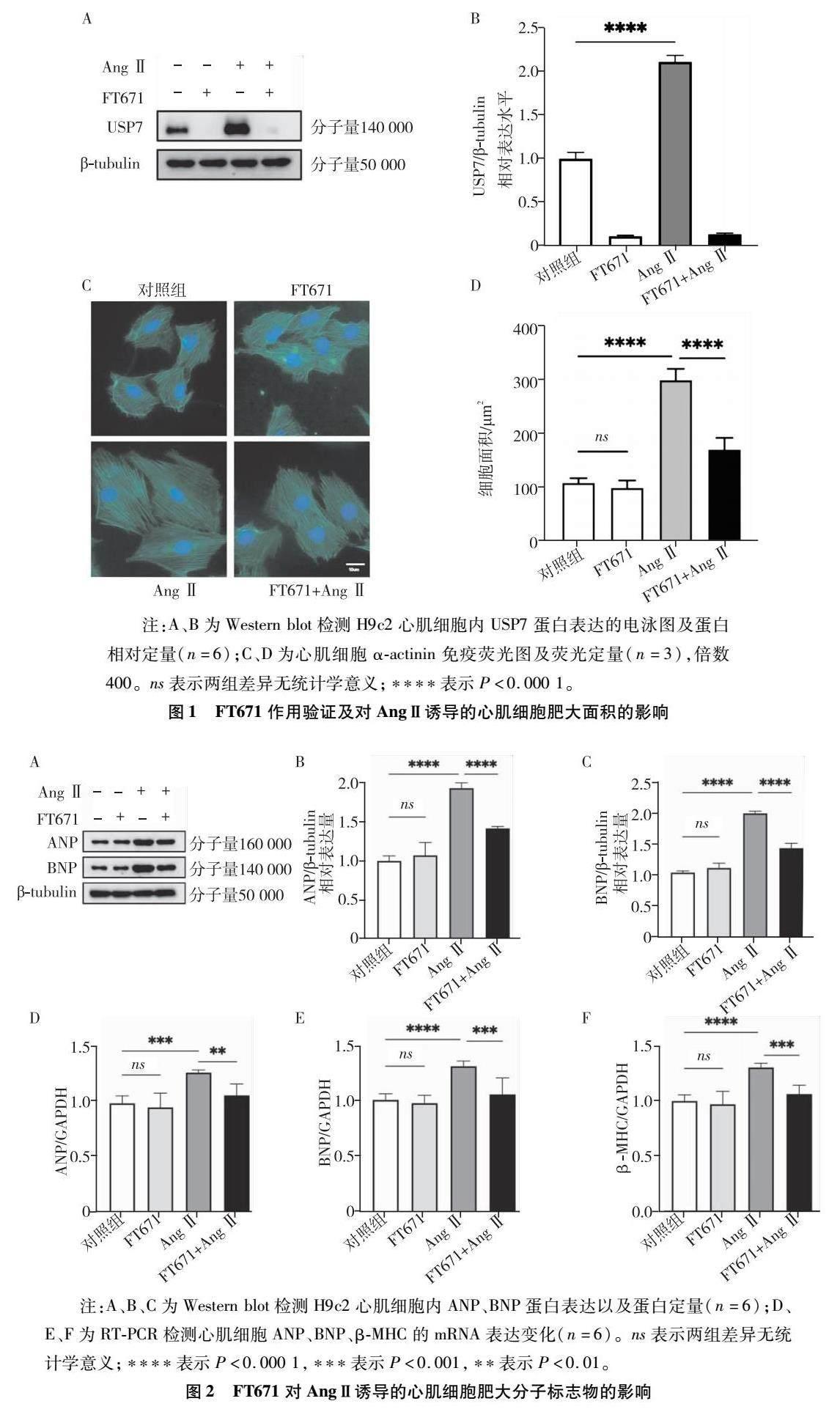
2.1??FT671作用驗(yàn)證及對(duì)AngⅡ誘導(dǎo)的心肌細(xì)胞肥大面積的影響
western blot檢測(cè)顯示AngⅡ刺激的心肌細(xì)胞中USP7蛋白表達(dá)明顯增加,在應(yīng)用USP7抑制劑FT671后,F(xiàn)T671組和FT671+AngⅡ組USP7表達(dá)明顯減少(圖1A、B)。對(duì)不同組H9C2細(xì)胞進(jìn)行α-actinin免疫熒光染色后,與空白對(duì)照組比,AngⅡ組和FT671+AngⅡ組心肌細(xì)胞面積增加,而相比于AngⅡ組,應(yīng)用FT671后心肌細(xì)胞面積明顯減少(圖1C、D),差異有統(tǒng)計(jì)學(xué)意義(P<0.05)。

注:A、B為western blot檢測(cè)H9C2心肌細(xì)胞內(nèi)USP7蛋白表達(dá)的電泳圖及蛋白相對(duì)定量(n=6);C、D為心肌細(xì)胞α-actinin免疫熒光圖及熒光定量(n=3),倍數(shù)400![]() 。ns表示兩組差異無(wú)統(tǒng)計(jì)學(xué)意義;****表示P<0.000?1。
。ns表示兩組差異無(wú)統(tǒng)計(jì)學(xué)意義;****表示P<0.000?1。
圖1?FT671作用驗(yàn)證及對(duì)AngⅡ誘導(dǎo)的心肌細(xì)胞肥大面積的影響
2.2??FT671對(duì)AngⅡ誘導(dǎo)的心肌細(xì)胞肥大分子標(biāo)志物的影響
應(yīng)用western blot檢測(cè)各組心肌細(xì)胞ANP、BNP的蛋白表達(dá)變化,real time-PCR檢測(cè)各組心肌細(xì)胞ANP、BNP、β-MHC的mRNA表達(dá)變化。結(jié)果顯示,AngⅡ干預(yù)組H9C2心肌細(xì)胞ANP、BNP蛋白(圖2A、B、C)以及ANP、BNP、β-MHC的mRNA(圖2D、E、F)的表達(dá)較對(duì)照組明顯增高(P<0.05),而FT671能降低AngⅡ誘導(dǎo)的ANP、BNP、β-MHC的表達(dá),差異具有統(tǒng)計(jì)學(xué)意義(P<0.05)。

注:A、B、C為western blot檢測(cè)H9C2心肌細(xì)胞內(nèi)ANP、BNP蛋白表達(dá)以及蛋白定量(n=6);D、E、F為real time-PCR檢測(cè)心肌細(xì)胞ANP、BNP、β-MHC的mRNA表達(dá)變化(n=6)。ns表示兩組差異無(wú)統(tǒng)計(jì)學(xué)意義;****表示P<0.000?1,***表示P<0.001,**表示P<0.01。
圖2 FT671對(duì)AngⅡ誘導(dǎo)的心肌細(xì)胞肥大分子標(biāo)志物的影響
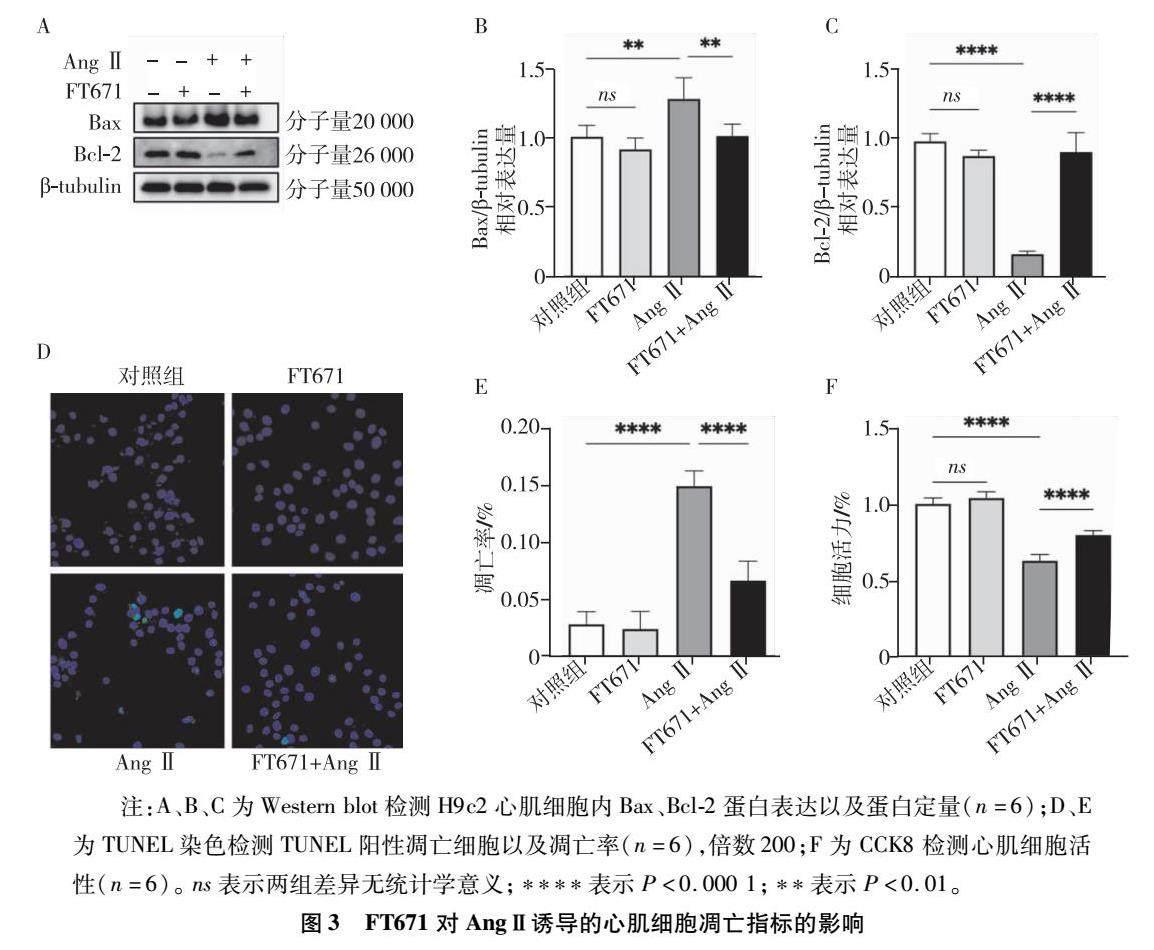
2.3??FT671對(duì)AngⅡ誘導(dǎo)的心肌細(xì)胞凋亡指標(biāo)的影響
應(yīng)用western blot檢測(cè)各組心肌細(xì)胞BCL-2和Bax的蛋白表達(dá)變化,結(jié)果顯示,與空白對(duì)照組比,AngⅡ組心肌細(xì)胞Bax表達(dá)增加,BCL-2表達(dá)降低,而相比于AngⅡ組,F(xiàn)T671+AngⅡ組心肌細(xì)胞Bax下調(diào),BCL-2表達(dá)上調(diào)(圖3A、B、C),差異有統(tǒng)計(jì)學(xué)意義(P<0.05)。TUNEL染色顯示相比于AngⅡ組,F(xiàn)T671組TUNEL陽(yáng)性細(xì)胞數(shù)明顯減少,心肌細(xì)胞凋亡率下降(圖3D、E),差異具有統(tǒng)計(jì)學(xué)意義(P<0.05)。應(yīng)用CCK8法檢測(cè)各組心肌細(xì)胞活性的變化,與空白對(duì)照組相比,F(xiàn)T671對(duì)H9C2心肌細(xì)胞活性的影響無(wú)統(tǒng)計(jì)學(xué)差異(P>0.05);而相比于AngⅡ組,F(xiàn)T671能明顯提高AngⅡ刺激后的細(xì)胞存活率(圖3F),差異具有統(tǒng)計(jì)學(xué)意義(P<0.05)。

注:A、B、C為western blot檢測(cè)H9C2心肌細(xì)胞內(nèi)Bax、BCL-2蛋白表達(dá)以及蛋白定量(n=6);D、E為TUNEL染色檢測(cè)TUNEL陽(yáng)性凋亡細(xì)胞以及凋亡率(n=6),倍數(shù)200![]() 。F為CCK8法檢測(cè)心肌細(xì)胞活性(n=6)。 ns表示兩組差異無(wú)統(tǒng)計(jì)學(xué)意義;****表示P<0.0001;**表示P<0.01。
。F為CCK8法檢測(cè)心肌細(xì)胞活性(n=6)。 ns表示兩組差異無(wú)統(tǒng)計(jì)學(xué)意義;****表示P<0.0001;**表示P<0.01。
圖3?FT671對(duì)AngⅡ誘導(dǎo)的心肌細(xì)胞凋亡指標(biāo)的影響
2.4??FT671對(duì)AngⅡ誘導(dǎo)的心肌細(xì)胞肥大的作用機(jī)制
既往研究表明,AngⅡ促進(jìn)心肌細(xì)胞內(nèi)ROS生成是其誘導(dǎo)心肌肥大的重要機(jī)制,因此筆者檢測(cè)了FT671對(duì)AngⅡ刺激的細(xì)胞內(nèi)ROS生成的影響,發(fā)現(xiàn) AngⅡ干預(yù)組ROS水平較對(duì)照組明顯升高,F(xiàn)T671干預(yù)可以明顯降低AngⅡ誘導(dǎo)的心肌細(xì)胞內(nèi)ROS的產(chǎn)生(P<0.05)(圖4A)。另外,筆者檢測(cè)了炎癥通路分子NLRP3[14]和ROS生成相關(guān)的關(guān)鍵酶Nox4[15]的蛋白表達(dá)水平。結(jié)果顯示,相比于AngⅡ組,F(xiàn)T671可以降低心肌細(xì)胞內(nèi)NLRP3和Nox4的蛋白表達(dá)水平(圖4B、C、D)。real time-PCR檢測(cè)顯示FT671干預(yù)可以降低AngⅡ誘導(dǎo)的IL-6、MCP-1、TNF-α炎性因子的表達(dá)(圖4E、F、G)。這些實(shí)驗(yàn)表明FT671通過(guò)抑制ROS介導(dǎo)的Nox4/NLRP3炎癥通路改善AngⅡ誘導(dǎo)的心肌細(xì)胞肥大。

注:A為ROS染色檢測(cè)心肌細(xì)胞內(nèi)ROS含量(n=6),倍數(shù)200![]() ;B、C、D為western blot檢測(cè)心肌細(xì)胞內(nèi)Nox4、NLRP3蛋白表達(dá)及蛋白定量(n=6);E、F、G為real time-PCR檢測(cè)心肌細(xì)胞IL-6、MCP-1、TNF-α的mRNA表達(dá)變化(n=6);?ns表示兩組差異無(wú)統(tǒng)計(jì)學(xué)意義;****表示P<0.000?1;***表示P<0.001;**表示P<0.01。
;B、C、D為western blot檢測(cè)心肌細(xì)胞內(nèi)Nox4、NLRP3蛋白表達(dá)及蛋白定量(n=6);E、F、G為real time-PCR檢測(cè)心肌細(xì)胞IL-6、MCP-1、TNF-α的mRNA表達(dá)變化(n=6);?ns表示兩組差異無(wú)統(tǒng)計(jì)學(xué)意義;****表示P<0.000?1;***表示P<0.001;**表示P<0.01。
圖4?FT671對(duì)AngⅡ誘導(dǎo)的心肌細(xì)胞肥大的作用機(jī)制
3??討論
在本研究中,證明了USP7抑制劑FT671在調(diào)節(jié)AngⅡ誘導(dǎo)的心肌細(xì)胞肥大中起著重要作用。本研究利用USP7抑制劑FT671從形態(tài)學(xué)和分子標(biāo)志物兩方面進(jìn)行了驗(yàn)證,發(fā)現(xiàn)USP7在AngⅡ刺激后的心肌細(xì)胞中表達(dá)顯著增加。FT671抑制USP7顯著減輕了AngⅡ誘導(dǎo)的心肌細(xì)胞內(nèi)肥大、凋亡、炎性反應(yīng)和ROS的產(chǎn)生。機(jī)制上,F(xiàn)T671可能是通過(guò)抑制Nox4相關(guān)的氧化應(yīng)激和NLRP3炎性小體的活化,改善心肌細(xì)胞肥大。總的來(lái)說(shuō),本研究發(fā)現(xiàn)USP7參與AngⅡ誘導(dǎo)的心肌肥大過(guò)程,并提示抑制USP7可能是治療心肌肥厚的一種潛在新靶點(diǎn)。
泛素化和去泛素化過(guò)程是一個(gè)動(dòng)態(tài)過(guò)程,調(diào)節(jié)特定的蛋白質(zhì)-蛋白質(zhì)相互作用。USP7是一種DUB,通過(guò)催化泛素分子與底物之間酰胺鍵的水解將泛素分子從標(biāo)記蛋白上水解下來(lái),阻止目標(biāo)蛋白被降解,從而調(diào)節(jié)細(xì)胞生理過(guò)程[16]。已知USP7可以調(diào)節(jié)MDM2/p53[17-18]、磷酸酶張力蛋白同源物(phosphatase and tensin homolog,PTEN)[19]、NLRP3[20]等靶蛋白的降解,在病毒復(fù)制、細(xì)胞信號(hào)轉(zhuǎn)導(dǎo)、DNA損傷修復(fù)、表觀遺傳調(diào)控和免疫反應(yīng)中發(fā)揮重要作用[21]。近年來(lái),USP7在心血管領(lǐng)域的研究逐漸增多,既往研究發(fā)現(xiàn),USP7在缺血再灌注損傷小鼠模型中可促進(jìn)心肌細(xì)胞炎癥和凋亡[13]。
本研究發(fā)現(xiàn)AngⅡ刺激的心肌細(xì)胞中USP7表達(dá)增加(圖1),表明 USP7 可能參與AngⅡ誘導(dǎo)的心肌細(xì)胞肥大過(guò)程。AngⅡ刺激后心肌細(xì)胞表面積明顯增加,肥厚標(biāo)志物ANP、BNP、β-MHC表達(dá)增加,而FT671可緩解上述肥厚基因的表達(dá)(圖1、2)。這表明靶向抑制USP7可能有助于減輕AngⅡ誘導(dǎo)的心肌肥厚。之前的研究[22-24]已經(jīng)證實(shí),凋亡,炎性反應(yīng)和氧化應(yīng)激在心肌梗死后心肌重構(gòu)中起著關(guān)鍵作用。因此筆者檢測(cè)并發(fā)現(xiàn)應(yīng)用FT671后AngⅡ刺激的心肌細(xì)胞中促凋亡因子Bax的增加減少,而抗凋亡因子BCL2表達(dá)明顯增加。除此之外,CCK8和TUNEL染色的結(jié)果也證明應(yīng)用FT671可以改善心肌細(xì)胞的存活率,減少細(xì)胞凋亡(圖3)。這些結(jié)果提示USP7參與調(diào)節(jié)AngⅡ誘導(dǎo)的心肌細(xì)胞凋亡過(guò)程。ROS生成過(guò)多會(huì)加劇心肌細(xì)胞內(nèi)的氧化損傷,推動(dòng)心肌細(xì)胞損傷和凋亡進(jìn)程[25]。Nox4是催化ROS生成的關(guān)鍵酶,下調(diào)Nox4的水平可以減少ROS的產(chǎn)生,減輕心肌細(xì)胞內(nèi)的氧化應(yīng)激水平,改善心肌肥大[26]。本研究顯示FT671的使用降低了Ang Ⅱ誘導(dǎo)的氧化應(yīng)激,這體現(xiàn)在其抑制ROS產(chǎn)生和Nox4的表達(dá)上(圖4)。更進(jìn)一步研究發(fā)現(xiàn),F(xiàn)T671抑制USP7后下調(diào)了NLRP3水平,減少了炎性因子IL-6、MCP-1、TNF-α的表達(dá)(圖4),這可能與NLRP3炎性小體受抑制有關(guān)系。
USP7在小鼠心肌缺血再灌注損傷過(guò)程中表達(dá)增加,抑制USP7表達(dá)可抑制氧自由基的生成和心肌細(xì)胞凋亡,減輕心肌組織損傷,改善心功能[27]。本研究證實(shí)抑制USP7可以減輕AngⅡ誘導(dǎo)的心肌細(xì)胞內(nèi)的凋亡和炎性反應(yīng),降低ROS的生成。Liu等[20]發(fā)現(xiàn)USP7可以靶向并去泛素化Nox4,從而導(dǎo)致ROS產(chǎn)生增加。本研究也證實(shí)了抑制USP7可以降低AngⅡ誘導(dǎo)的Nox4水平。另外。有研究[28]證明USP7可以調(diào)節(jié)NLRP3炎性小體的泛素化狀態(tài)。本研究也發(fā)現(xiàn)抑制USP7可以下調(diào)心肌細(xì)胞中NLRP3的水平。因此,USP7可能通過(guò)靶向Nox4和NLRP3蛋白發(fā)揮去泛素化作用,從而參與AngⅡ誘導(dǎo)的炎性反應(yīng)和氧化應(yīng)激過(guò)程,調(diào)節(jié)心肌肥厚和心肌重構(gòu)。本研究仍存在一些局限性。目前,AngⅡ誘導(dǎo)USP7表達(dá)增加的具體機(jī)制尚不清楚,需要在未來(lái)進(jìn)一步探索。
總之,本研究表明,在AngⅡ刺激的心肌細(xì)胞中,USP7表達(dá)增加,并且通過(guò)靶向Nox4和NLRP3的去泛素化來(lái)調(diào)節(jié)AngⅡ誘導(dǎo)的心肌細(xì)胞內(nèi)的氧化應(yīng)激和炎性反應(yīng)從而參與心肌肥厚的形成,而抑制USP7或可成為治療心肌重構(gòu)中心肌肥厚的新靶點(diǎn)。
利益沖突 所有作者均聲明不存在利益沖突
參考文獻(xiàn)
[1]??McDonagh TA,Metra M,Adamo M,et al.2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure[J].Eur Heart J,2021,42(36):3599-3726.
[2] Xie S,Chen M,F(xiàn)ang W,et al. Diminished arachidonate 5-lipoxygenase perturbs phase separation and transcriptional response of Runx2 to reverse pathological ventricular remodeling[J].EBioMedicine,2022,86:104359.
[3] Swatek KN,Komander D. Ubiquitin modifications[J].Cell Res,2016,26(4):399-422.
[4] Zheng J,Chen C,Guo C,et al.The pleiotropic ubiquitin-specific peptidase 16 and its many substrates[J].Cells,2023,12(6):886.
[5] Clague MJ,Urbé S,Komander D. Breaking the chains: deubiquitylating enzyme specificity begets function[J].Nat Rev Mol Cell Biol,2019,20(6):338-352.
[6] Hu Y,Ma Z,Chen Z,et al.USP47 promotes apoptosis in rat myocardial cells after ischemia/reperfusion injury via NF-kappaB activation[J].Biotechnol Appl Biochem,2021,68(4):841-848.
[7] Han X,Zhang YL,F(xiàn)u TT,et al.Blockage of UCHL1 activity attenuates cardiac remodeling in spontaneously hypertensive rats[J].Hypertens Res,2020,43(10):1089-1098.
[8] Bi HL,Zhang YL,Yang J,et al. Inhibition of UCHL1 by LDN-57444 attenuates Ang II-Induced atrial fibrillation in mice[J].Hypertens Res,2020,43(3):168-177.
[9] Pozhidaeva A,Bezsonova I. USP7: structure,substrate specificity,and inhibition[J].DNA Repair (Amst),2019,76:30-39.
[10] Fountain MD,Oleson DS,Rech ME,et al. Pathogenic variants in USP7 cause a neurodevelopmental disorder with speech delays,altered behavior,and neurologic anomalies[J].Genet Med,2019,21(8):1797-1807.
[11] Zhang XW,F(xiàn)eng N,Liu YC,et al. Neuroinflammation inhibition by small-molecule targeting USP7 noncatalytic domain for neurodegenerative disease therapy[J].Sci Adv,2022,8(32):eabo0789.
[12] Galarreta A,Valledor P,Ubieto-Capella P,et al. USP7 limits CDK1 activity throughout the cell cycle[J].EMBO J,2021,40(11):e99692.
[13] Xue Q,Yang D,Zhang J,et al. USP7,negatively regulated by miR-409-5p,aggravates hypoxia-induced cardiomyocyte injury[J].APMIS,2021,129(3):152-162.
[14] Suetomi T,Willeford A,Brand CS,et al. Inflammation and NLRP3 inflammasome activation initiated in response to pressure overload by Ca2+/calmodulin-dependent protein kinase Ⅱ delta signaling in cardiomyocytes are essential for adverse cardiac remodeling[J].Circulation,2018,138(22):2530-2544.
[15] Bedard K,Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology[J].Physiol Rev,2007,87(1):245-313.
[16] Nie L,Wang C,Liu X,et al. USP7 substrates identified by proteomics analysis reveal the specificity of USP7[J].Genes Dev,2022,36(17-18):1016-1030.
[17] Meulmeester E,Maurice MM,Boutell C,et al.Loss of HAUSP-mediated deubiquitination contributes to DNA damage-induced destabilization of Hdmx and Hdm2[J].Mol Cell,2005,18(5):565-276.
[18] Cummins JM,Rago C,Kohli M,et al.Tumour suppression: disruption of HAUSP gene stabilizes p53[J].Nature,2004,428(6982):1 p following 486.
[19] Song MS,Salmena L,Carracedo A,et al. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network[J].Nature,2008,455(7214):813-817.
[20] Liu G,Liu Q,Yan B,et al.USP7 inhibition alleviates H2O2-induced injury in chondrocytes via inhibiting NOX4/NLRP3 pathway[J].Front Pharmacol,2020,11:617270.
[21] Ji L,Lu B,Zamponi R,et al. USP7 inhibits Wnt/beta-catenin signaling through promoting stabilization of Axin[J].Nat Commun,2019,10(1):4184.
[22] Sanz RL,Inserra F,García Menéndez S,et al.Metabolic syndrome and cardiac remodeling due to mitochondrial oxidative stress involving gliflozins and sirtuins[J].Curr Hypertens Rep,2023;25(6):91-106.
[23] Daou D,Gillette TG,Hill JA. Inflammatory mechanisms in heart failure with preserved ejection fraction[J].Physiology (Bethesda),2023,38(5):0.
[24] Hu C,Zhang X,Wei W,et al. Matrine attenuates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via maintaining AMPKalpha/UCP2 pathway[J].Acta Pharm Sin B,2019,9(4):690-701.
[25] Werbner B,Tavakoli-Rouzbehani OM,F(xiàn)atahian AN,et al.The dynamic interplay between cardiac mitochondrial health and myocardial structural remodeling in metabolic heart disease,aging,and heart failure[J].J Cardiovasc Aging,2023,3(1):9.
[26] Guo S,Chen X. The human Nox4:gene,structure,physiological function and pathological significance[J].J Drug Target,2015,23(10):888-896.
[27] Xu Q,Liu M,Gu J,et al. Ubiquitin-specific protease 7 regulates myocardial ischemia/reperfusion injury by stabilizing Keap1[J].Cell Death Discov,2022,8(1):291.
[28] Palazón-Riquelme P,Worboys JD,Green J,et al. USP7 and USP47 deubiquitinases regulate NLRP3 inflammasome activation[J].EMBO Rep,2018,19(10):e44766.
收稿日期:2023-05-04
猜你喜歡
中成藥(2021年5期)2021-07-21 08:39:04
世界科學(xué)技術(shù)-中醫(yī)藥現(xiàn)代化(2020年2期)2020-07-25 02:05:56
中成藥(2018年6期)2018-07-11 03:01:24
中成藥(2018年5期)2018-06-06 03:11:43
天然產(chǎn)物研究與開(kāi)發(fā)(2016年6期)2016-06-05 10:29:26
西南軍醫(yī)(2016年6期)2016-01-23 02:21:19
新疆醫(yī)科大學(xué)學(xué)報(bào)(2015年10期)2015-12-26 12:33:30
吉林大學(xué)學(xué)報(bào)(醫(yī)學(xué)版)(2015年4期)2015-12-17 07:48:13
實(shí)用中西醫(yī)結(jié)合臨床(2015年7期)2015-02-28 16:30:23
癌變·畸變·突變(2015年3期)2015-02-27 06:15:09
