玉米多葉矮化突變體lyd1的鑒定與基因克隆
2024-04-28 01:13:44劉孝偉牛群凱時子文侯雨微馮開潔榮廷昭曹墨菊
作物學報 2024年5期
蘇 帥 劉孝偉 牛群凱 時子文 侯雨微 馮開潔 榮廷昭 曹墨菊
玉米多葉矮化突變體的鑒定與基因克隆
蘇 帥 劉孝偉 牛群凱 時子文 侯雨微 馮開潔 榮廷昭 曹墨菊*
四川農業大學玉米研究所 / 農業部西南玉米生物學與遺傳育種重點實驗室, 四川成都 611130
玉米株高降低通常是由節間數目減少、節間長度變短或二者共同作用所致。而本研究在基因編輯后代中發現的玉米多葉矮化突變體卻表現為節間數目顯著增加, 株高顯著降低。株高僅為93.10 cm, 與野生型KN5585的株高159.95 cm相比, 降低了41.79%, 差異達到極顯著水平。然而葉片數平均達到27.8片, 相較野生型平均17.8片葉, 增加56.18%, 差異達到極顯著水平。遺傳分析表明,的突變表型由1對隱性核基因控制, 通過圖位克隆將控制多葉矮化性狀的基因定位于玉米3號染色體標記Indel10和Indel11之間, 物理距離0.74 Mb。對定位區間內13個基因(不包含假基因)的序列進行測序, 發現僅在第4外顯子出現1個A堿基的替換, 其他基因無差異。編碼一個RNA結合蛋白, 氨基酸的替換發生在第3個RNA結合結構域內(RRM3), 導致天冬氨酸轉變為纈氨酸。突變體的突變位點與已報道的、、、不同,的發現為進一步解析玉米葉片和節間發育平衡的遺傳機制提供了寶貴的材料。
玉米; 葉片數量; 節間長度; 基因定位
玉米(L.)是我國糧食和飼料作物, 在保障國家糧食安全方面發揮重要作用。現階段玉米產量的增長主要通過提升種植密度, 適當降低株高以增強抗倒伏能力是提高種植密度的關鍵[1-2]。玉米節間數量(葉片數量)和節間長度是決定株高的關鍵因素, 因此對于玉米葉片數量和節間長度以及協調二者平衡相關基因的挖掘具有重要的理論和實踐意義。
玉米節間長度由節間居間分生組織細胞的分裂、分化和伸長決定。植物激素作為重要的信號分子, 參與玉米節間長度的調控。[3]、[4-5][6]編碼赤霉素合成途徑的中間產物,[7-9]、[9]編碼DELLA蛋白作為GA信號轉導的負調控轉錄因子, 通過調控赤霉素的生物合成或信號轉導影響玉米節間長度。生長素以極性運輸的方式對節間長度產生影響,編碼生長素極性運輸蛋白PGP, 通過促進節間生長素的積累抑制節間的伸長, 且對穗下部節間長度影響較大[10-11]。[12]、[13]位于油菜素內酯生物合成的上游,[14]位于油菜素內酯生物合成的下游,參與油菜素內酯信號轉導途徑[15], 上述基因突變后均會對玉米節間長度產生影響。編碼乙烯生物合成中的關鍵限速酶ACC合成酶, 通過增強蛋白的穩定性, 促進乙烯合成, 抑制節間伸長[16]。玉米葉片由莖頂端分生組織的周緣區以一定的時間間隔和空間間隔分化形成[17], 但具體的分子機制尚未解析。迄今已經報道的葉片數量相關突變體主要與生長素和細胞分裂素有關。玉米基因編碼IAA生物合成途徑中的色氨酸轉移酶,突變體展示出葉片數目顯著減少而導致的矮化表型[18]。編碼細胞分裂素誘導的A型反應調節因子, 玉米突變體葉片數增多, 但對株高沒有顯著影響。在突變體中, 玉米IAA外排載體在初始葉原基的表達量大幅下降, 其生長素水平也明顯降低[19]。
RNA結合蛋白是含有一個或多個RNA結合結構域, 通過結合RNA并改變結合RNA的命運或功能的蛋白質[20-21]。RNA結合蛋白的研究主要在哺乳動物和細菌系統中進行, 植物RNA結合蛋白研究較少。是在酵母中發現的一個編碼RNA結合蛋白的基因, 含有3個RNA結合結構域, 促進酵母細胞減數分裂S期DNA的合成和減數第一次分裂的有序進行[22]。水稻基因編碼Mei2-like蛋白, 該基因突變后植株矮小, 葉片數增加, 植株早熟[23]。是在玉米中的同源基因, 該基因在葉原基起始處表達量最低, 其突變體株高降低, 葉片數目增多, 葉脈紊亂, 植株雄穗缺失, 原雄穗發育處展示出雌性化的特征[33-34]。盡管在植物中很多基因已經被發現, 且均與酵母基因類似, 含有3個RNA結合結構域, 但在植物中未找到酵母下游基因的同源物, 表明該基因在酵母和植物中的功能和作用機制并不相同[24]。在植物中基因下游結合的RNA的挖掘, 以及3個RNA結合結構域在發揮功能過程中的作用仍需要進一步深入研究。
本研究以多葉矮化突變體()為研究對象, 對突變體的葉片數和節間長度等表型特征進行了系統調查, 采用分子標記技術對突變基因進行了定位, 并對候選基因進行預測和分析, 確定了關鍵候選基因, 該基因編碼RNA結合蛋白。本研究為進一步闡明玉米葉片數量和節間長度的平衡關系以及RNA結合蛋白對玉米生長發育的影響提供了寶貴材料。
1 材料與方法
1.1 試驗材料
玉米多葉矮化突變體、玉米自交系B73、Mo17和KN5585。突變體選自基因()的CRISPR/Cas9敲除后代, 對突變體中的進行測序, 結果顯示突變體中未發生編輯, 即該突變體的突變表型與無關。
1.2 突變體lyd1的表型鑒定和遺傳分析
2020年6月在四川農業大學溫江試驗基地發現該多葉矮化突變體, 將其自交保種。2020年12月在四川農業大學云南試驗基地種植、Mo17和B73, 并將分別與Mo17和B73雜交得到F1。2021年3月在四川農業大學溫江試驗基地種植F1, 自交獲得F2種子。2022年3月在四川農業大學溫江試驗基地種植F2群體。成熟期統計F2分離群體中正常和多葉矮化植株的分離比并進行卡平方測驗。
2021年3月將野生型KN5585和突變體種植于四川農業大學溫江試驗基地, 野生型KN5585二葉期后, 每間隔3 d, 統計野生型KN5585和突變體各30株完全展開葉數量。在植株成熟期, 田間隨機選取野生型KN5585和各15株, 考察株高、穗位、葉片長度、葉片寬度、雄穗分支數、地上部分各節間長度和散粉期等重要農藝性狀。使用Microsoft Excel 2016進行數據處理和繪圖。
1.3 莖稈細胞學觀察
植株散粉后, 選取野生型KN5585和地上部分最長節間(均為雌穗下方第2節, 野生型KN5585為第11節, 突變晰完體為第19節)的中部, 縱切并將其固定于FAA固定液中。將樣品依次進行軟化、洗滌、脫水、透明、包埋、切片、烤片、染色, 在顯微鏡下進行節間細胞觀察, 野生型KN5585和突變體分別選取細胞形態清整的3張切片, 共10個視野進行照相。通過ImageJ軟件進行細胞長度測量, 并根據實際測量得出的最長節間平均長度進行節間細胞數目的計算, 使用Microsoft Excel 2016進行數據處理和繪圖。
1.4 基因定位
利用CTAB法[25]提取玉米自交系Mo17、KN5585、突變體以及[×Mo17] F2分離群體內多葉矮化單株的基因組DNA。利用均勻分布在玉米10條染色體上1050對Indel和SSR標記, 對Mo17和進行多態性分析。在[×Mo17] F2群體中, 選取10株多葉矮化單株提取葉片的DNA, 稀釋成相同濃度并等體積混合, 每5株混合為一個池, 構建2個突變池。同時, 另外選取10株表型正常的單株提取葉片的DNA, 稀釋成相同濃度并等體積混合為一個正常池。采用BSA (bulk segregant analysis)方法[26]篩選在正常池和突變池之間具有穩定性差異的多態性標記, 并利用上述標記對F2群體內多葉矮化單株進行基因分型, 篩選與突變表型連鎖的多態性標記, 對目標基因進行初步定位。利用NCBI在線引物設計網站根據初步定位區段內序列信息, 設計產物片段600 bp左右的引物, 通過PCR擴增出親本基因片段后進行測序, 利用DNAMAN軟件對所擴增的雙親基因序列進行比對分析, 找出雙親間的序列差異位點, 根據差異位點開發片段大小為150~300 bp的Indel標記。利用新開發的標記, 對擴大的定位群體內多葉矮化單株進行基因分型, 縮小定位區間, 對目的基因進行精細定位。
1.5 候選基因的預測與分析
在MaizeGDB和NCBI網站上, 獲取精細定位區間內基因的注釋信息。根據精細定位區間內基因序列和注釋, 設計基因特異引物, 以野生型和基因組DNA或全長CDS為模板, 利用KOD高保真DNA聚合酶進行PCR擴增。擴增體系為30 μL: 2×PCR buffer for KOD FX 15 μL、正向和反向引物(10 μmol L–1)各2 μL、KOD FX 0.6 μL、dNTPs (0.002 mol L–1) 6 μL、ddH2O 3.4 μL、模板3 μL, PCR程序為: 95℃預變性5 min; 98℃變性10 s, 58℃退火30 s, 68℃延伸(1 kb min–1), 循環數為35次; 68℃ 10 min。PCR擴增產物送公司進行測序并利用DNAMAN軟件進行序列比對, 確定變異位點。
以定位群體內關鍵交換單株DNA為模板, 根據變異位點附近序列設計特異性引物, 對變異位點所在的基因片段進行擴增, 長度約600 bp, 變異位點位于擴增片段中間, 并將PCR擴增產物送公司進行測序, 利用DNAMAN軟件進行序列比對, 進行變異位點群體內共分離驗證。
1.6 關鍵候選基因的表達及其蛋白的亞細胞定位
為了檢測在不同時期不同組織的表達模式, 利用TRIzol試劑盒提取野生型KN5585在V3、V7、V11、V17時期的根、莖、葉、雌穗、雄穗的RNA; 同時為了檢測突變體中基因表達水平變化, 利用TRIzol試劑盒提取野生型KN5585和的V3和V7時期莖尖的RNA, 電泳檢測RNA的完整性, 并將質量符合標準的RNA反轉錄得到cDNA, 根據玉米公共數據庫MaizeGDB中提供的候選基因cDNA信息, 使用Primer 3在線設計特異性引物, 引物擴增片段長度在100~200 bp左右。cDNA稀釋至100 ng μL–1作為模板。以作為內參。qRT-PCR反應在CFX 96 Real-Time System上進行。qRT-PCR反應體系為10 μL: Diluted cDNA 1 μL, 2×SYBR Green PCR Mix 5 μL, 上游引物(10 μmol L–1) 0.4 μL, 下游引物(10 μmol L–1) 0.4 μL, ddH2O 3.2 μL。反應程序為: 95℃預變性3 min; 95℃變性10 s, 58℃退火30 s, 72℃延伸15 s, 40個循環, 3次生物重復, 3次技術重復。擴增結束后產生的數據導入Microsoft Excel中, 采用2–DDCt方法進行基因相對表達量的分析。
為了檢測蛋白在細胞內的定位模式, 設計帶有PCAMBIA2300-eGFP載體同源臂的引物擴增全長轉錄本。利用同源重組的方法, 將pCAMBIA2300-35S-eGFP載體和目的片段連接, 并轉入大腸桿菌感受態, 挑選陽性克隆檢測并測序比對, 挑選正確序列的單克隆擴繁, 提取質粒。將質粒轉化GV3101農桿菌, 200轉 min–128℃過夜擴繁至OD600為0.6左右, 然后5000轉 min–1離心5 min收集菌體, 棄上清, 加入適量的注射液。用已滅菌的1 mL注射器將菌體懸浮液注射至4~8周左右的煙草葉背部表皮細胞層中, 然后在正常條件下培養48 h, 用熒光顯微觀察并采集圖像。
2 結果與分析
2.1 突變體的表型鑒定及遺傳分析
為了明確突變體的遺傳規律, 將自交后代于每年的春季在四川、秋季在云南進行多年種植觀察,的突變表型不受環境影響, 可以穩定遺傳。將分別與Mo17和B73雜交, F1代植株均表現正常表型, 將F1自交獲得F2, 在5380株[×Mo17] F2群體中, 正常植株數量為4083株, 多葉矮化株數量為1297株, 經卡平方檢測, 正常植株與多葉矮化株分離比符合3∶1 (c2C=2.24, 小于c20.05,1=3.84); 在286株[×B73] F2群體中, 正常植株數量為220株,多葉矮化株數量為66株, 經卡平方檢測, 正常植株與多葉矮化株分離比符合3∶1 (c2C=0.46, 小于c20.05,1=3.84)。不同遺傳背景下的F2群體, 正常植株與多葉矮化植株分離比均符合3∶1, 表明該突變表型是由單個隱性核基因控制。
對突變體的農藝性狀進行調查發現,株高93.10 cm、穗位高43.48 cm, 僅為野生型株高159.95 cm、穗位高64.37 cm的58.21%和67.55% (圖1-A~C), 進一步分析表明穗上部和穗下部節間縮短是造成株高降低的原因(圖2-A)。生育期內完全葉數量平均為27.80±1.18片, 野生型生育期內完全葉數量平均為(17.80±0.95)片(圖2-B), 差異達到極顯著水平。野生型從播種至群體內50%植株散粉需要(89.03±2.10) d,從播種至群體內50%植株散粉需要(88.64±1.79) d, 二者無顯著差異(表1)。綜合葉片數量及生育期的比較結果可以發現,相鄰2片葉片完全展開的時間間隔為2.31 d, 野生型相鄰2片葉片完全展開的時間間隔為3.50 d, 兩者差異達到顯著水平,相鄰葉原基起始所用時間間隔縮短, 葉片起始速率加快。此外葉片長度和寬度較野生型顯著縮小(圖1-D), 穗長、穗粗、行粒數、穗粒重、百粒重均顯著下降(圖1-F, G), 雄穗分支數顯著減少, 雄穗基部由未完全伸展的葉片包裹(圖1-E和表1), 這可能也暗示了雄穗基部節間伸長受阻。

圖1 野生型KN5585和突變體lyd1的性狀比較
A、B、C: 分別表示五葉期、十葉期和成熟期的野生型KN5585和植株; D: 野生型KN5585和成熟期的葉片表型; E: 野生型KN5585和突變體的雄穗表型; F、G: 野生型KN5585和的果穗和籽粒的表型。標尺: A 15 cm; B、C、D 30 cm; E 5 cm; F 2 cm。
A, B, and C: represent the 5th leaf stage, 10th leaf stage, and mature stage of wild type KN5585 and mutant, respectively. D: leaves phenotypes of wild type KN5585 and mutantat maturity. E: the tassel phenotype of wild type KN5585 and mutant. F, G: phenotypes of ear and kernel of wild types KN5585 and mutant. Bar: A 15 cm; B, C, D 30 cm; E 5 cm; F 2 cm.

圖2 野生型KN5585和突變體lyd1的節間、葉片發育的比較
A: 野生型KN5585和地上部分節間長度的比較, 0表示雌穗所在節間、–1表示雌穗下方第一節、+1表示雌穗上方第一節, 依此類推; B: 野生型KN5585和葉片發育過程比較。縱坐標表示調查群體內植株葉片的平均數, 橫坐標表示調查日期。
A: the comparison of internodes length between the wild type KN5585 and mutant, 0 represents the internode of the ear, –1 represents the first internode below the ear, +1 represents the first internode above the ear, and so on. B: the comparison of leaves development between wild type KN5585 and mutant. The vertical axis shows the average number of leaves in the surveyed population. The horizontal axis indicates the survey date.
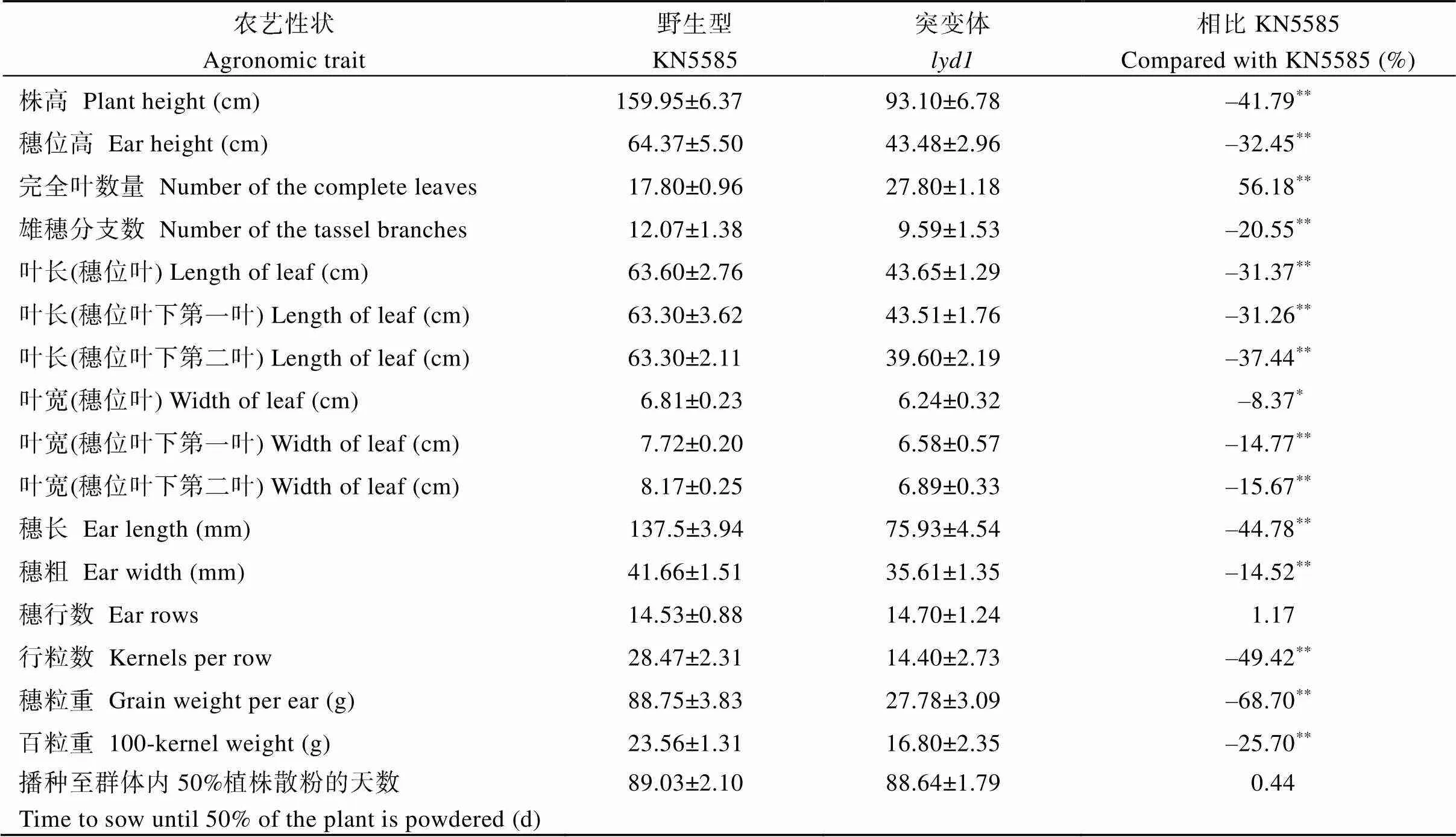
表1 突變體lyd1和野生型KN5585的農藝性狀數據分析
表中數據以“平均值±標準差”形式呈現;**表示< 0.01差異極顯著,*表示< 0.05差異顯著。
Data are present “means ± SDs” in the table;**:< 0.01;*:< 0.05.
對和野生型最長節間進行石蠟切片觀察(圖3-A, B)。結果表明,最長節間的10個細胞縱向長度為17.70 μm, 與野生型KN5585最長節間的10個細胞縱向長度22.13 μm相比, 減少了20.02%, 差異達到極顯著水平;最長節間細胞數目36,003.50個, 野生型KN5585最長節間細胞數目44,984.00個, 差異達到極顯著水平(圖3-C, D)。因此,節間縮短是由于節間細胞數目減少和細胞縱向長度縮短共同導致。

圖3 野生型KN5585和突變體lyd1散粉后雌穗穗位下第二節節間的石蠟切片觀察
A: 散粉后野生型KN5585最長節間(第11葉所在節間)細胞觀察; B: 散粉后突變體最長節間(第19葉所在節間)細胞觀察; C, D: 野生型KN5585最長節間和最長節間細胞長度和數量的比較。標尺: 5 μm;**表示< 0.01差異極顯著。
A: the observation of longest intersegmental (the internode where the 11th leaf is located.) cells of wild type KN5585 after loose powder; B: the observation of longest intersegmental (the internode where the 19th leaf is located.) cells of mutantafter loose powder; C, D: the comparison of longest intersegmental cell length and number between wild type KN5585 and mutant. Bar: 5 μm;**:< 0.01
2.2 突變基因的定位
將[×Mo17] F2作為定位群體, 利用均勻分布在玉米10條染色體上的SSR和Indel標記對、Mo17進行多態性分析, 從中共篩選到172對具有雙親多態性的標記。將篩選得到的多態性標記對正常基因池和突變基因池進行BSA混池分析, 結果顯示位于3號染色體上3對多態性標記Bnlg1019、Indel1、Indel9 (表2)在正常基因池和突變基因池之間存在差異。進一步利用[×Mo17] F2中的93株多葉矮化單株進行基因分型, 發現標記Bnlg1019、Indel1和Indel9的交換單株分別為35、34和9個, 因此將候選基因定位于玉米3號染色體標記Indel1和Indel9之間。在Indel1和Indel9之間開發出8對新的多態性Indel標記, 利用8對Indel標記對[×Mo17] F2中93個多葉矮化單株進行基因分型, 發現Indel2、Indel3、Indel4、Indel5、Indel6、Indel7、Indel10、Indel8 (表2)的交換單株數分別為30、15、10、7、6、2、0和8個, 將定位區段鎖定在標記Indel7和Indel8之間, 物理距離10.94 Mb (圖4-A)。
為了精細定位, 在標記Indel7和Indel8之間新開發出5對有多態性的標記。同時擴大定位群體, 利用新開發的5對多態性標記對[×Mo17] F2群體中的剩余的1204株多葉矮化單株進行基因分型, Indel7、Indel10、SNP1、Indel11、Indel12、Indel13和Indel8 (表2)的交換單株數分別為35、6、0、11、11、39和89個, 最終將候選基因定位在Indel10和Indel11標記之間, 物理距離約0.74 Mb (圖4-B, C)。
2.3 候選基因分析
根據B73_V5版本的基因注釋信息, 0.74 Mb定位區間中共有14個候選基因, 其中存在1個假基因(表3)。根據候選基因的基因組和CDS序列的長度及擴增的難易程度, 對基因組序列較短且較容易擴增的基因擴增其基因組序列, 否則擴增其CDS序列, 并將擴增產物進行測序。結果顯示KN5585和之間在基因組上、、、、、和均無序列差異, 在全長CDS上、、、、均無序列差異。而()在基因組上存在1個堿基的替換, 為了驗證該堿基替換的真實性, 對全長CDS序列進行擴增, 發現該位置的堿基替換在的基因組和CDS序列中穩定存在(圖5-A)。全長4132 bp, 只有1個轉錄本, 包含6個外顯子和5個內含子, 編碼664個氨基酸, 具有3個典型的RNA結合結構域(RRM), 編碼一個RNA結合蛋白。中堿基替換發生在第4外顯子上, 位于全長CDS第1478堿基由A堿基轉換為T堿基, 導致第492個氨基酸由天冬氨酸轉變為纈氨酸, 該突變的氨基酸殘基位于RRM3結構域內。RRM3在玉米、水稻、擬南芥、小麥、高粱、大豆、藜麥等植物中保守性較高, 并且突變的天冬氨酸也具有較高保守性(圖5-B, C)。
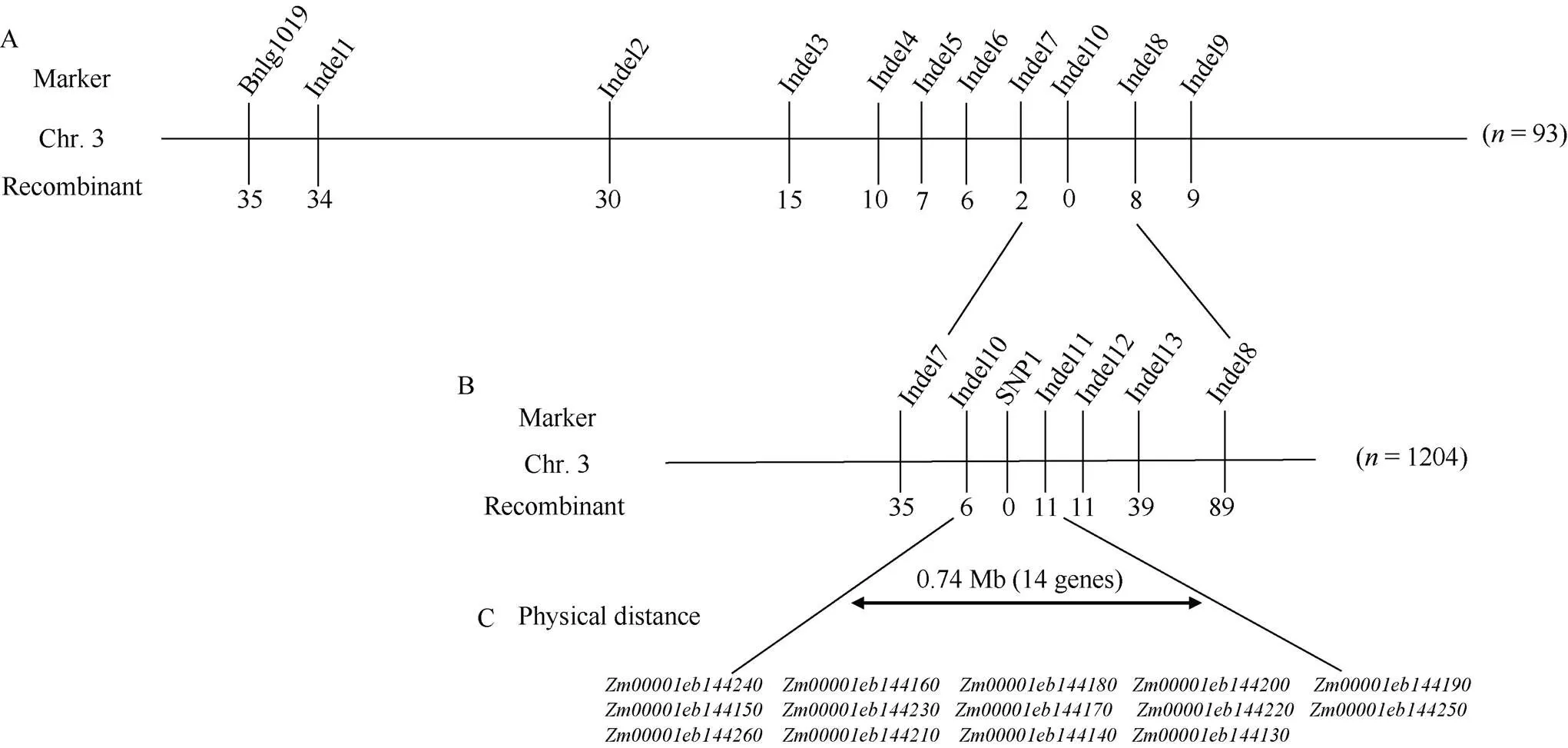
圖4 突變體lyd1的精細定位
A: 候選基因初步定位在3號染色體Indel7和Indel8分子標記之間; B: 候選基因精細定位在3號染色體Indel10和Indel11分子標記之間; C: 精細定位區間內包含14個候選基因。橫線上方為分子標記, 橫線下方為重組單株數。
A: the candidate gene was preliminarily located between the Indel7 and Indel8 molecular markers on chromosome 3; B: the candidate gene was finely positioned between the Indel10 and Indel11 molecular markers on chromosome 3; C: there are 14 candidate genes in the fine localization interval. Above the horizontal line is the molecular marker, below the horizontal line is the number of recombinant individual plants.
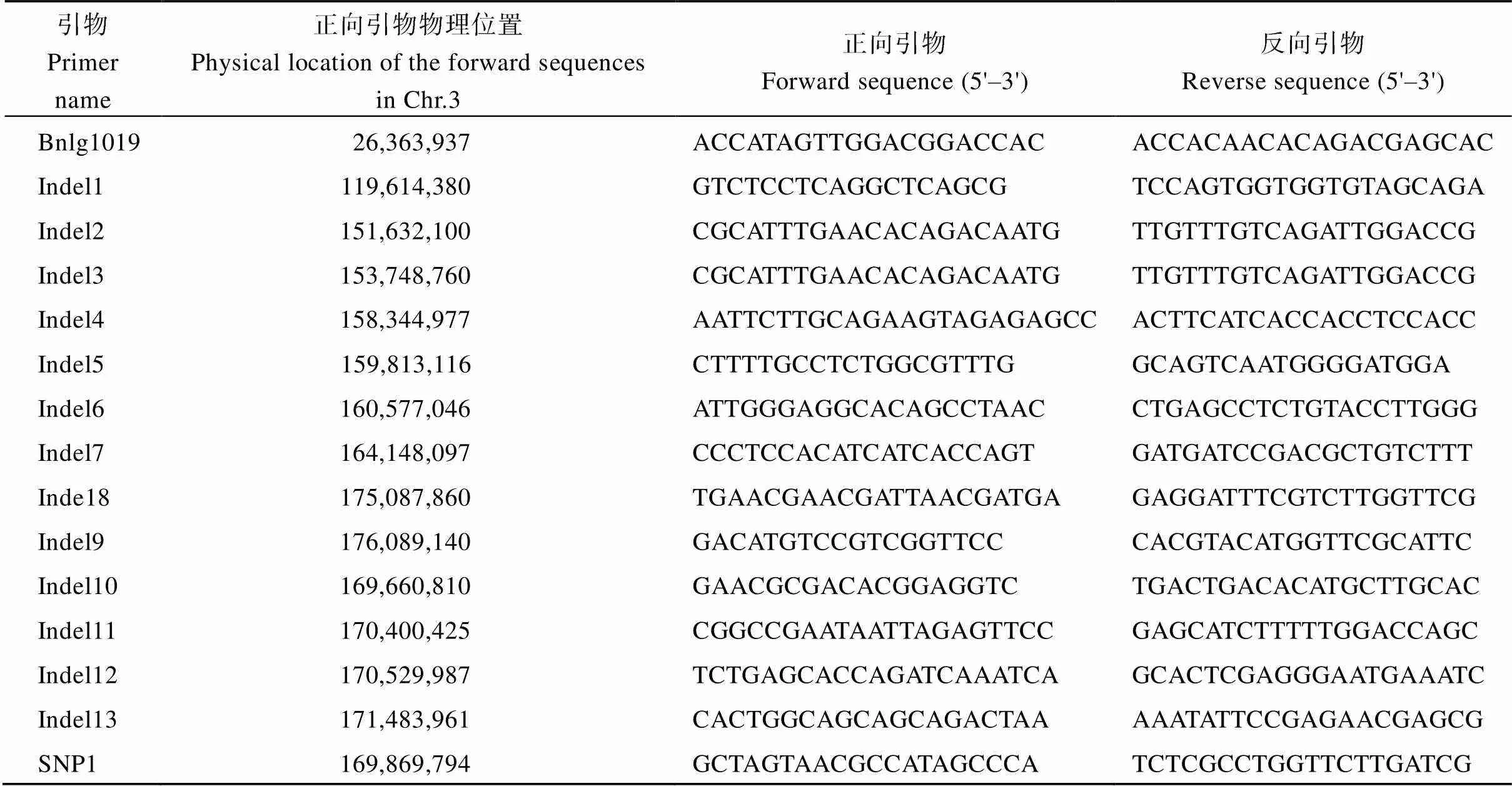
表2 基因定位相關標記信息
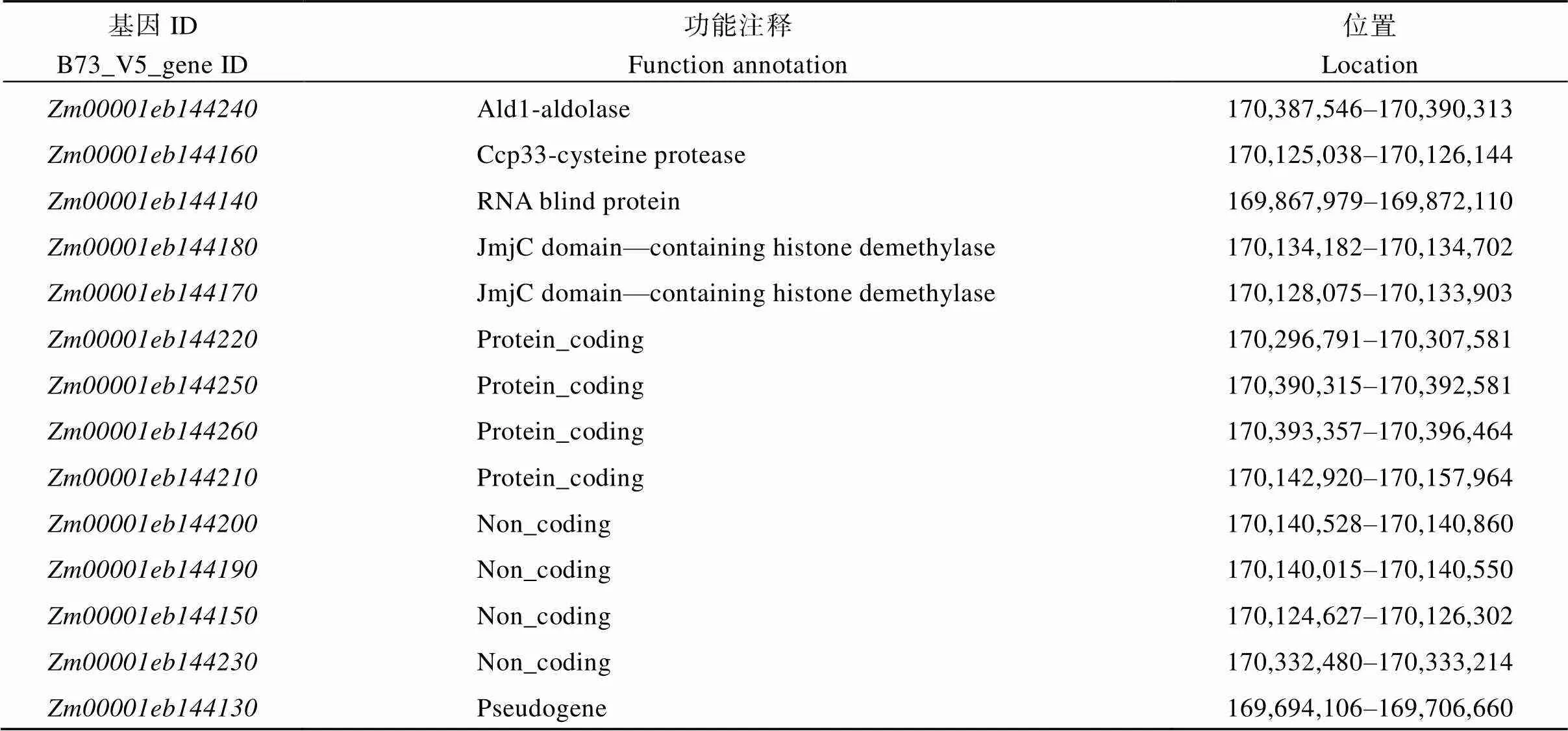
表3 定位區間內候選基因功能注釋
根據在和野生型差異位點設計特異性SNP引物(SNP1), 以標記Indel10和Indel11在[×Mo17] F2定位群體中檢測到的17個關鍵交換單株的基因組DNA為模板, 進行擴增測序, 測序結果顯示17個單株在該位點都顯示的T堿基(圖6), 以上結果表明該位置發生的堿基替換與多葉矮化突變表型共分離。
2.4 ZmTE1的表達分析
利用qRT-PCR分析了在野生型的表達特征, 結果顯示在莖尖和幼嫩的雌穗中表達量較高, 在葉片、莖、雄穗中表達量很低, 在根中幾乎不表達(圖7-A)。同時分析了V3和V7時期在野生型與莖尖中的表達情況, 結果顯示2個時期在突變體和野生型之間的表達量均未達顯著差異(圖7-B), 說明A堿基到T堿基的替換可能不影響該基因在莖尖的表達。
為進一步了解蛋白的定位情況, 對蛋白進行亞細胞定位分析, 結果顯示GFP-的GFP信號與核、膜Marker的信號很好地重疊(圖8-A), 表明蛋白定位在細胞核和細胞膜上, 這與RNA結合蛋白在轉錄水平對細胞核內的mRNA進行加工的功能一致。
3 討論
玉米的株高通常由節間的數目和節間長度決定, 一般來說節間數目(葉片數目)越多, 節間越長植株越高。目前已報道的玉米矮化突變體, 有些葉片數目不變, 株高降低由節間長度縮短導致, 如[10-11]、[27]、[7-9]、[28]過表達株系[29]等, 有些為葉片數目減少和節間長度縮短, 但葉片數減少是導致株高降低的主要原因, 如[18]、過表達株系[30]等。此外還有些玉米矮化突變體, 表現為節間數量的增加, 其株高降低是由節間長度縮短導致, 如突變體葉片數較野生型增加3.2片[31];突變體葉片數較野生型增加1.6片[32];突變體葉片數較野生型增加1.75片[16]。而本研究發現的多葉矮化突變體葉片數較野生型KN5585增加10片葉, 增加了56.18%, 節間嚴重縮短, 為研究節間數目和節間長度的平衡提供了寶貴材料。
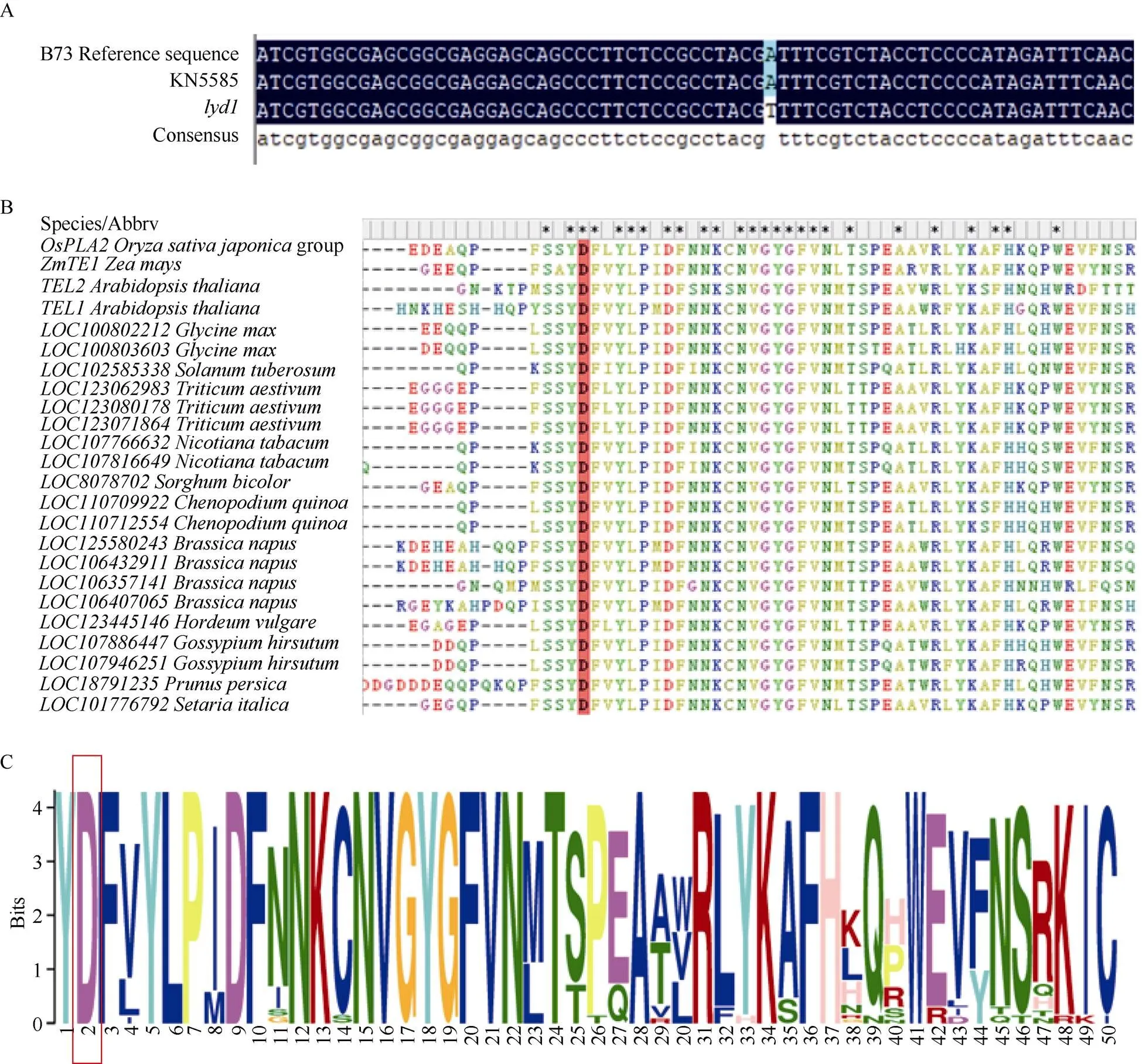
圖5 lyd1突變位點及突變位點保守性分析
A: 突變體與野生型KN5585的CDS差異序列比較; B, C:蛋白序列突變氨基酸殘基保守性分析
A: the comparison of CDS difference sequence ofin mutantand wild type KN5585; B, C: the conservative analysis of amino acid residues mutated inprotein sequence.
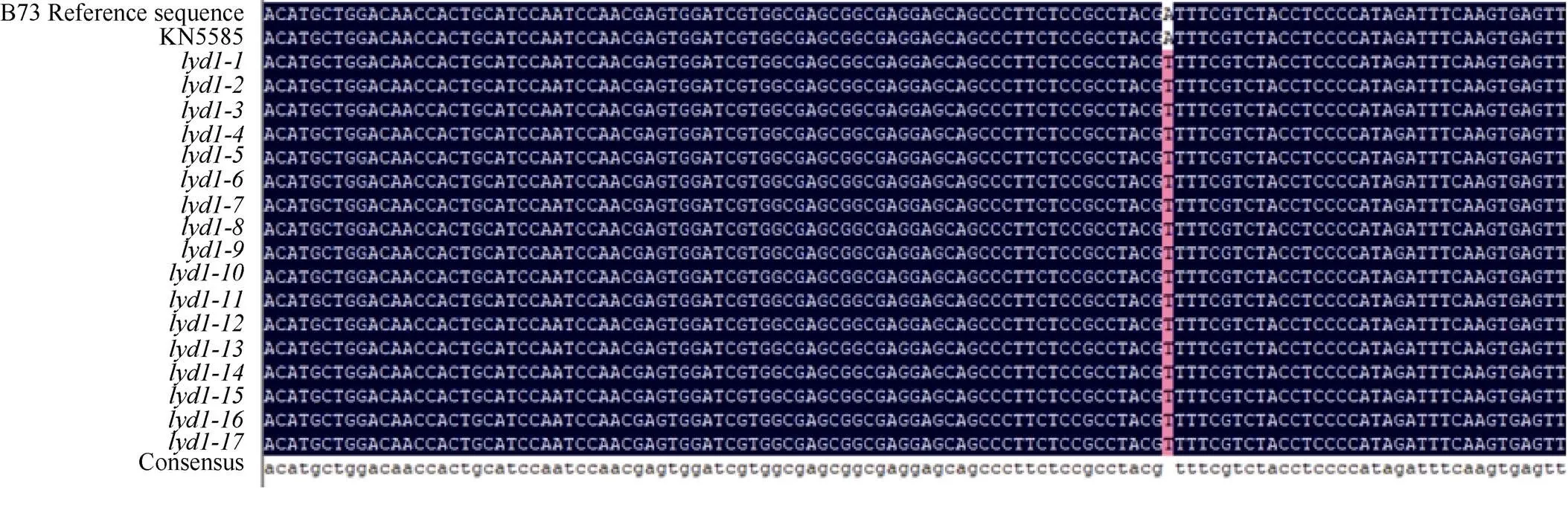
圖6 野生型KN5585和突變體lyd1的序列差異位點群體驗證
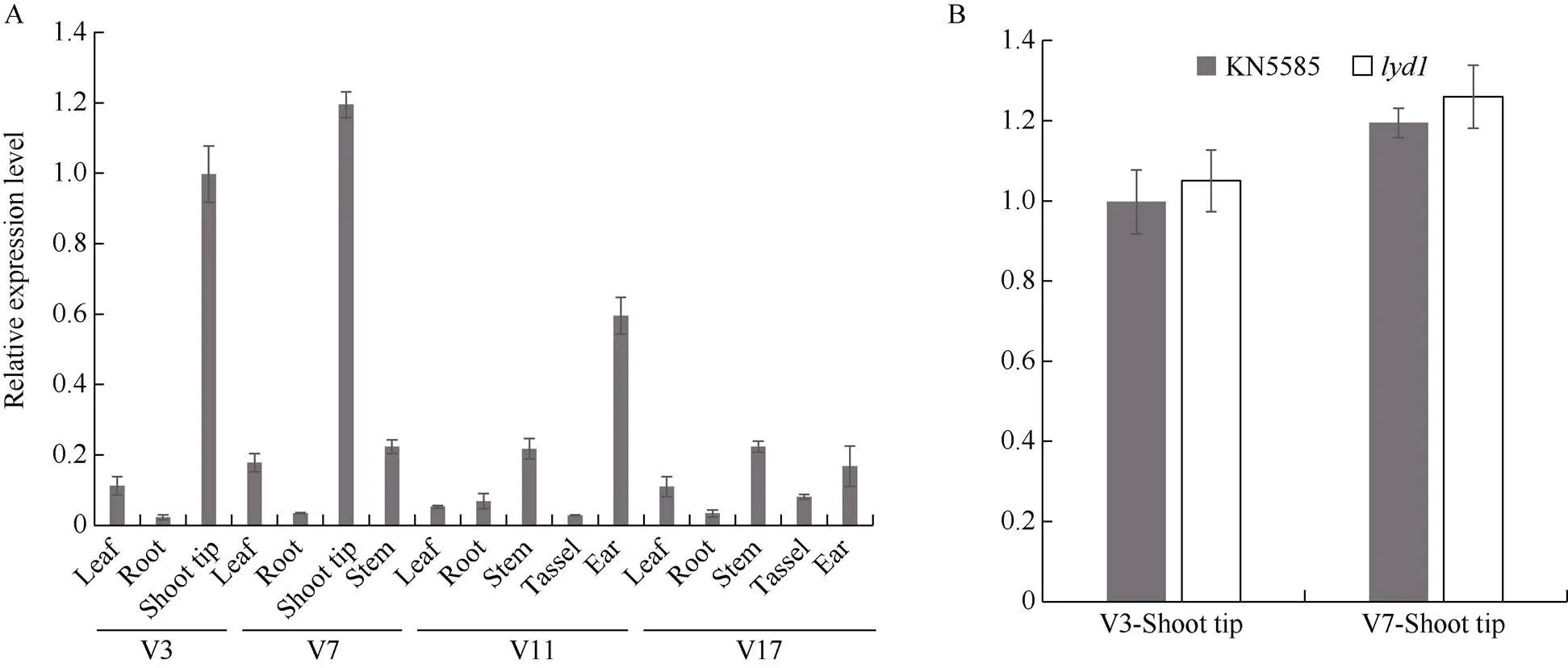
圖7 ZmTE1組織表達分析
A:在野生型KN5585不同組織中的表達; B:在野生型KN5585和突變體中的表達比較。V3、V7、V11、V17分別表示玉米植株分別有3、7、11、17片完全展開葉所處的時期。
A: the relative expression level ofgenes in different tissues of wild type KN5585; B: the comparison ofexpression in wild type KN5585 and mutant. V3, V7, V11, and V17 represent the stage when the maize has 3, 7, 11, and 17 fully unfolded leaves, respectively.
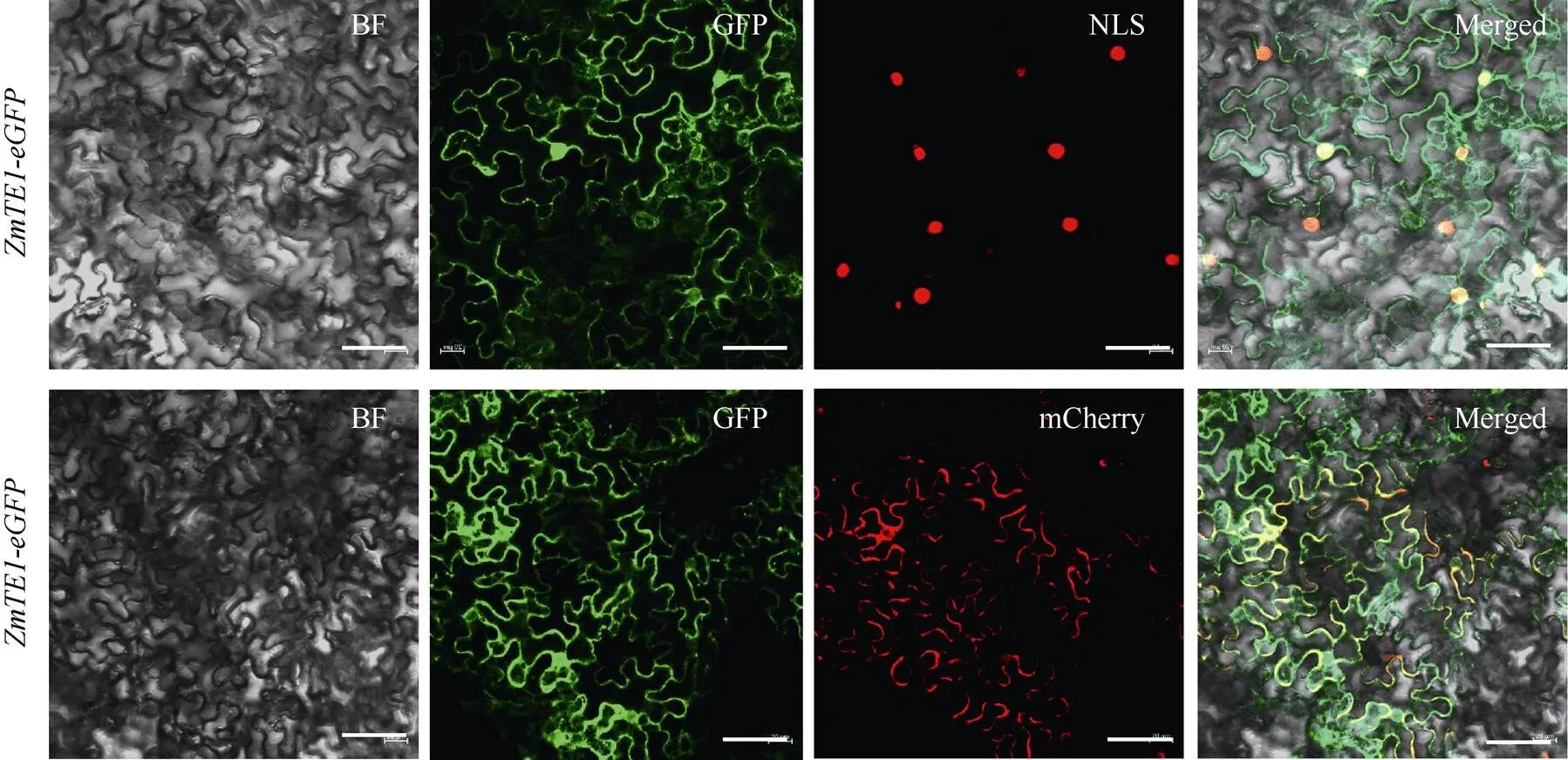
圖8 ZmTE1的亞細胞定位
PC2300-eGFP-與NLS (細胞核標記)和mCherry (細胞膜標記)共定位, 標尺為50 μm。
PC2300-eGFP-are colocalized with NLS (nuclear marker) and mCherry (cell membrane marker). Bar: 50 μm.
控制突變表型的基因被定位在玉米3號染色體, 位于標記Indel10和Indel11之間, 物理距離0.74 Mb, 將確定為的關鍵候選基因。與已報道的、、突變體的突變位點不同,和突變體中轉座子插入在第3外顯子,突變體中轉座子插入在第1內含子[33],突變體中第1外顯子單堿基的替換導致蛋白翻譯提前終止[34], 上述已報道的突變體植株生活力較弱, 難以完成正常生長發育, 給后續研究帶來困難, 并且由于突變位點的不同導致株高降低的程度存在差異, 突變體僅在第4外顯子上發生1處堿基替換, 植株矮化程度較輕, 且并未出現雌穗和雄穗發育缺失, 能夠正常生長。RNA結合蛋白通過在轉錄水平上改變與之結合RNA的命運或功能, 調控植物的生長發育[35]。RRM是特征最明確的RNA結合基序, 其拓撲結構為βαββαβ, 與RNA的特異性識別通常發生在β-sheet的表面[36]。編碼含有3個RRM結構域的RNA結合蛋白, 但該基因下游的靶標RNA尚未找到。突變體中發生突變的天冬氨酸殘基具有較高的保守性, 且出現在保守性較高的RRM3結構域內, 位于與RNA的特異性識別相關的β-sheet的表面, 因此我們推測中的點突變可能通過影響與下游RNA的結合能力進而影響其他基因的表達。由此可見,突變體的發現對于從RRM3結構域出發探索下游靶標RNA, 以及解析3個RRM在多葉矮化表型產生過程中所扮演的角色具有重要研究意義。
4 結論
本研究發現了一個玉米多葉矮化突變體, 其株高較野生型KN5585顯著下降41.79%, 葉片數較野生型KN5585顯著增加56.18%。該突變表型受一對隱性核基因控制, 被定位在3號染色體分子標記Indel10和Indel11之間, 物理距離0.74 Mb。進一步對定位區間內候選基因測序, 發現第4外顯子出現1個堿基替換, 其他基因無差異, 因此, 將確定為突變表型的關鍵候選基因, 該基因編碼一個含有3個RNA結合結構域的RNA結合蛋白, 突變位點位于第3個RNA結合結構域。突變體的發現為探究玉米葉片數目(節間數目)與節間長度的平衡以及二者對株型發育的影響提供了寶貴的試驗材料。
[1] Tester M, Langridge P. Breeding technologies to increase crop production in a changing world., 2010, 327: 818–822.
[2] Haarhoff S J, Swanepoel P A. Plant population and maize grain yield: a global systematic review of rainfed trials., 2018, 5: 1819–1829.
[3] Bensen R J, Johal G S. Cloning and characterization of the maizegene., 1995, 7: 75–84.
[4] Chen Y, Hou M M, Liu L J, Wu S, Shen Y, Ishiyama K, Kobaya, Shi M, McCarty D R, Tan B C. The maizeencodes a gibberellin 3-oxidase and is dual localized to the nucleus and cytosol., 2014, 166: 2028–2039.
[5] Teng F, Zhai L H, Liu R X, Bai W, Wang L Q, Huo D G, Tao Y S, Zheng Y L, Zhang Z X., a candidate gene for a major QTL,, for plant height in maize., 2013, 73: 405–416.
[6] Winkler R G, Helentjaris T. The maizegene encodes a cytochrome P450-mediated early step in gibberellin biosynthesis., 1995, 7: 1307–1317.
[7] Cassani E, Bertolini E, Cerino Badone F, Landoni M, Gavina D, Sirizzotti A, Pilu R. Characterization of the first dominant dwarf maize mutant carrying a single amino acid insertion in the VHYNP domain of thegene., 2009, 24: 375–385.
[8] Harberd N P, Freeling M. Genetics of dominant gibberellin- insensitive dwarfism in maize., 1989, 121: 827–838.
[9] Lawit S J, Wych H M, Xu D, Kundu S, Tomes D T. Maize DELLA proteinsandas modulators of plant development., 2010, 51: 1854–1868.
[10] Multani D S, Briggs S P, Chamberlin M A, Blakeslee J J, Murphy A S, Johal G S. Loss of an MDR transporter in compact stalks of maizeand sorghummutants., 2003, 302: 81–84.
[11] Zhang X, Hou X, Liu Y, Zheng L, Yi Q, Zhang H, Huang X, Zhang J, Hu Y, Yu G, Liu H, Li Y, Huang H, Zhan F, Chen L, Tang J, Huang Y. Maize brachytic2 () suppresses the elongation of lower internodes for excessive auxin accumulation in the intercalary meristem region., 2019, 19: 589.
[12] Hartwig T, Chuck G S, Fujioka S, Klempien A, Weizbauer R, Potluri D P V, Choe S, Johal G S, Schulz B. Brassinosteroid control of sex determination in maize., 2011, 108: 19814–19819.
[13] Best N B, Hartwig T, Budka J, Fujioka S, Johal G, Schulz B, Dilkes B P.encodes a maize ortholog of thebrassinosteroid biosynthesis gened, identifying developmental interactions between brassinosteroids and gibberellins., 2016, 171: 2633–2647.
[14] Makarevitch I, Thompson A, Muehlbauer G J, Springer N M.gene in maize encodes a brassinosteroid C-6 oxidase., 2012, 7: e30798.
[15] Kir G, Ye H, Nelissen H, Neelakandan A K, Kusnandar A S, Luo A, Inzé D, Sylvester A W, Yin Y, Becraft P W. RNA interference knock down ofin maize reveals novel functions for brassinosteroid signaling in controlling plant architecture., 2015, 169: 826–839.
[16] Li H, Wang L, Liu M, Dong Z, Li Q, Fei S, Xiang H, Liu B, Jin W. Maize plant architecture is regulated by the ethylene biosynthetic gene., 2020, 183: 1184–1199.
[17] Schaller G E, Bishopp A, Kieber J J. The Yin-Yang of hormones: cytokinin and auxin interactions in plant development., 2015, 27: 44–63.
[18] Phillips K A, Skirpan A L, Liu X, Christensen A, Slewinski T L, Hudson C, Barazesh S, Cohen J D, Malcomber S, Mcsteen P.encodes a grass-specific tryptophan aminotransferase required for vegetative and reproductive development in maize., 2011, 23: 550–566.
[19] Lee B H, Johnston R, Yang Y, Gallavotti A, Kojima M, Traven?olo B A, Costa Lda F, Sakakibara H, Jackson D. Studies ofmutants of maize indicate complex interactions between auxin and cytokinin signaling in the shoot apical meristem., 2009, 150: 205–216.
[20] 張在寶, 李婉杰, 李九麗, 張弛, 胡夢輝, 程琳, 袁紅雨. 植物RNA結合蛋白研究進展. 中國農業科學, 2018, 51: 4007–4019. Zhang Z B, Li W J, Li J L, Zhang C, Hu M H, Cheng L, Yuan H Y. The research progress of plant RNA binding proteins., 2018, 51: 4007–4019 (in Chinese with English abstract).
[21] Cho H, Cho H S, Hwang I. Emerging roles of RNA-binding proteins in plant development., 2019, 51: 51–57.
[22] Jeffares D C, Phillips M J, Moore S, Veit B. A description of the Mei2-like protein family; structure, phylogenetic distribution and biological context., 2004, 214: 149–158.
[23] Kawakatsu T, Itoh J, Miyoshi K, Kurata N, Alvarez N, Veit B, Nagato Y.regulates leaf initiation and maturation in rice., 2006, 18: 612–625.
[24] Anderson G H, Alvarez N D, Gilman C, Jeffares D C, Trainor V C, Hanson M R, Veit B. Diversification of genes encoding mei2–like RNA binding proteins in plants.,2004, 54: 653–670.
[25] 王關林, 方宏筠. 植物基因工程(第2版). 北京: 科學出版社, 2002. pp 742–744. Wang G L, Fang H Y. Plant Gene Engineering, 2nd edn. Beijing: Science Press, 2002. pp 742–744 (in Chinese).
[26] Michelmore R W, Paran I, Kesseli R V. Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations., 1991, 88: 9828–9832.
[27] Avila L M, Cerrudo D, Swanton C, Lukens L., a putative inositol polyphosphate 5-phosphatase, is required for internode elongation in maize., 2016, 67: 1577–1588.
[28] Zhang D, Sun W, Singh R, Zheng Y, Cao Z, Li M, Lunde C, Hake S, Zhang Z.regulates shoot architecture and meristem determinacy in maize., 2018, 30: 360–374.
[29] Li W, Ge F, Qiang Z, Zhu L, Zhang S, Chen L, Wang X, Li J, Fu Y. Maizeencodes a microtubule-associated protein that controls plant and ear height., 2020, 18: 1345–1347.
[30] Heuer S, Hansen S, Bantin J, Brettschneider R, Kranz E, L?rz H, Dresselhaus T. The maize MADS box geneaffects node number and spikelet development and is co-expressed withduring flower development, in egg cells, and early embryogenesis., 2001, 127: 33–45.
[31] Lyu H K, Zheng J, Wang T Y, Fu J J, Huai J L, Min H W, Zhang X, Tian B H, Shi Y S, Wang G Y. The maize, a novel allele of, is required for maize internode elongation., 2014, 84: 243–257.
[32] Bommert P, Je B I, Goldshmidt A, Jackson D. The maize Gα genefunctions in clavata signalling to control shoot meristem size., 2013, 502: 555–558.
[33] Veit B, Briggs S P. Regulation of leaf initiation by thegene of maize., 1998, 393: 166–168.
[34] Wang F, Yu Z, Zhang M, Wang M, Lu X, Liu X, Li Y, Zhang X, Tan B C, Li C, Ding Z.promotes plant height by regulating intercalary meristem formation and internode cell elongation in maize., 2022, 20: 526–537.
[35] Hentze M W, Castello A, Schwarzl T, Preiss T. A brave new world of RNA-binding proteins., 2018, 19: 327–341.
[36] 唐蜻. 植物RNA結合蛋白的研究進展. 安徽農業科學, 2010, 38(1): 38–41. Tang Q. The research progress of plant RNA binding proteins., 2010, 38(1): 38–41 (in Chinese with English abstract).
Identification and gene cloning of leafy dwarf mutantin maize
SU Shuai, LIU Xiao-Wei, NIU Qun-Kai, SHI Zi-Wen, HOU Yu-Wei, FENG Kai-Jie, RONG Ting-Zhao, and CAO Mo-Ju*
Maize Research Institute, Sichuan Agricultural University / Key Laboratory of Biology and Genetic Improvement of Maize in Southwest Region, Ministry of Agriculture and Rural Affairs, Chengdu 611130, Sichuan, China
The decrease of plant height in maize is usually caused by the decrease in the number of internodes, the shortening of internodes or the combination of both. However, in this study, the mutant() found in the progeny of gene editing, exhibited more leaves and shorter stature. Quantitative measurements indicated the plant height of mutantwas only 93.10 cm, the plant height of wild-type KN5585 was 159.95 cm. The plant height was significantly reduced by 41.79% in mutantcompared with the wild type KN5585. The wild type KN5585 produced an average of 17.8 leaves at maturity stage, whereas mutantsproduced 27.8 leaves. The number of leaves were significantly increased by 56.18% in mutantcompared with the wild type. Genetic analysis showed that the mutation phenotype ofwas controlled by a pair of recessive nuclear genes. We applied a map-based cloning strategy to identify the gene responsible for thephenotype. The gene was located between Indel10 and Indel11 on maize chromosome 3, and the physical distance was 0.74 Mb. Gene sequencing analysis of 13 genes (excluding pseudogenes) within the interval revealed that one base A was substituted in the fourth exon of, and there was no significant difference in other genes.encoded an RNA-binding protein. The amino acid substitution was in the third RNA binding domain (RRM3), resulting in the conversion of aspartic acid to valine. The mutation sites of the mutantwere different from,,, andin previously reported. The discovery ofprovides valuable materials for further analysis of the genetic mechanism of the balance between leaves and internodes development in maize.
maize; the number of leaves; the length of internodes; gene mapping
10.3724/SP.J.1006.2024.33044
本研究由四川省科技計劃項目(2021YFYZ0011, 2021YFYZ0017, MZGC20230108)和四川農業大學學科建設專項研究支持計劃項目資助。
This study was supported by the Sichuan Science and Technology Program (2021YFYZ0011, 2021YFYZ0017, MZGC20230108) and the Specific Research Supporting Program for Discipline Construction at Sichuan Agricultural University.
曹墨菊, E-mail: caomj@sicau.edu.cn
E-mail: 1018714902@qq.com
2023-07-31;
2024-01-12;
2024-02-08.
URL: https://link.cnki.net/urlid/11.1809.S.20240206.1114.002
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
