鎂基固態(tài)儲(chǔ)氫材料的研究進(jìn)展
2024-04-02 06:45:26梁宸曦王振斌張明錦馬存花
儲(chǔ)能科學(xué)與技術(shù) 2024年3期
梁宸曦,王振斌,2,張明錦,2,馬存花,2,梁 寧
(1青海師范大學(xué)化學(xué)化工學(xué)院,青海 西寧 810016;2青海省人民政府-北京師范大學(xué)高原科學(xué)與可持續(xù)發(fā)展研究院,青海 西寧810008;3河南省地質(zhì)礦產(chǎn)勘查開發(fā)局第三地質(zhì)勘查院,河南鄭州 451450;4河南省金屬礦產(chǎn)深孔鉆探工程技術(shù)研究中心,河南 鄭州 450003)
20 世紀(jì)以來,煤、石油、天然氣三大傳統(tǒng)化石能源大規(guī)模消耗和大量溫室氣體的產(chǎn)生,促使科研人員對可替代能源進(jìn)行了深入研究[1]。氫能作為一種理想的綠色能源,具有來源廣泛、燃燒熱值高(33.3 kWh/kg)、環(huán)境友好等優(yōu)異特性,在可持續(xù)能源體系的發(fā)展與過渡階段起著重要的作用[2]。氫可以從各種各樣的過程中產(chǎn)生,如水的電解、化石燃料的等離子弧分解、水的熱分解、生物質(zhì)轉(zhuǎn)化、光催化、生物光解等[3-7]。但氫氣易燃易爆,且密度低,易逸出,對儲(chǔ)存和運(yùn)輸帶來不便。因此,儲(chǔ)氫技術(shù)已成為當(dāng)前氫能應(yīng)用的關(guān)鍵步驟之一[8]。目前氫的存儲(chǔ)方式主要包括以下幾類:高壓氣態(tài)儲(chǔ)氫[9]、液態(tài)儲(chǔ)氫[10],以及固態(tài)儲(chǔ)氫[11]。各種儲(chǔ)氫方式的儲(chǔ)氫原理和優(yōu)缺點(diǎn)等具體細(xì)節(jié)見表1。
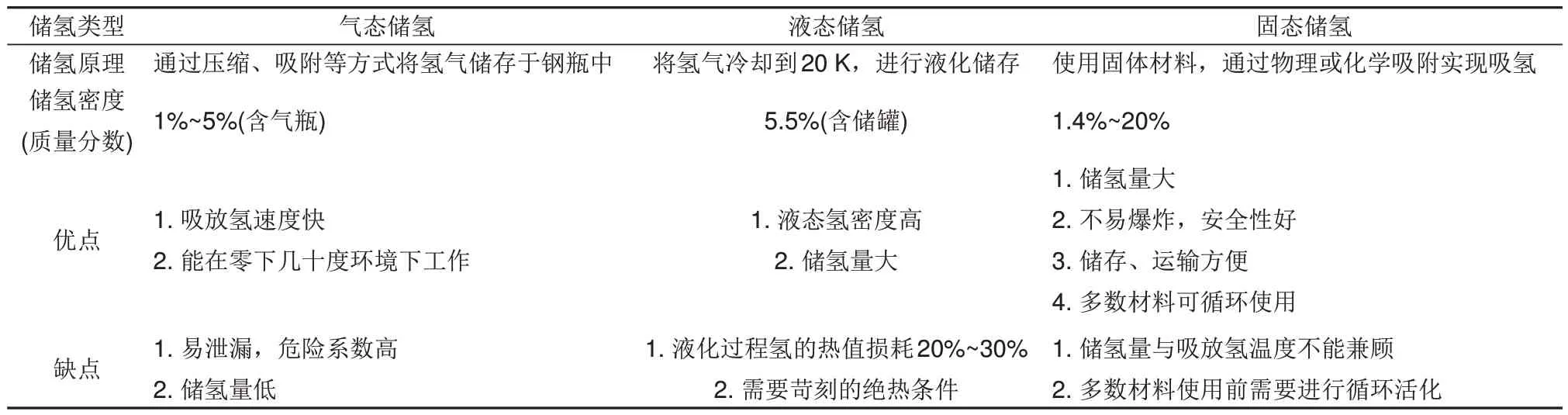
表1 氣、液、固態(tài)儲(chǔ)氫方式的比較Table 1 Comparison of gas, liquid and solid hydrogen storage methods
氣態(tài)儲(chǔ)氫可以儲(chǔ)存約4%(質(zhì)量分?jǐn)?shù))的氫氣,但壓縮過程充滿著危險(xiǎn)性,對容器耐壓要求較高。液氫的密度為70.8 kg/m3,使用液態(tài)儲(chǔ)氫系統(tǒng)可在一定程度上提高氫氣儲(chǔ)量,但液化過程中能耗高,對容器絕熱性要求較高[12-13]。理想的儲(chǔ)氫材料應(yīng)具有高儲(chǔ)氫密度、快速吸/放氫性能和長周期循環(huán)穩(wěn)定性。國際能源署(International Energy Agency,IEA)提出理想儲(chǔ)氫材料的性能標(biāo)準(zhǔn)為:質(zhì)量儲(chǔ)氫密度>5.5%(質(zhì)量分?jǐn)?shù)),放氫溫度<423 K,循環(huán)壽命>1000 次[14-15]。與氣態(tài)高壓和液態(tài)儲(chǔ)氫系統(tǒng)相比,固態(tài)儲(chǔ)氫材料的儲(chǔ)氫密度和安全性較高,具有滿足IEA目標(biāo)的巨大潛力。固態(tài)儲(chǔ)氫材料根據(jù)吸附劑和吸附質(zhì)之間作用力的不同可分為物理吸附和化學(xué)吸附兩種方式。前者包括傳統(tǒng)的碳基多孔材料、介孔材料、金屬有機(jī)框架(MOFs)等。物理吸附主要通過相對較弱的范德華力實(shí)現(xiàn)儲(chǔ)氫,吸附壓強(qiáng)較高且只能在較低溫度(77 K)下實(shí)現(xiàn)儲(chǔ)氫[16]。與前者不同的是,化學(xué)吸附通過較強(qiáng)的化學(xué)鍵實(shí)現(xiàn)儲(chǔ)氫,化學(xué)儲(chǔ)氫材料包括化學(xué)氫化物(Mg-N-H、NH3BH3、Li-NH)[17-19]、絡(luò)合氫化物(LiAlH4、LiBH4、NaBH4)[20-22]和金屬氫化物(MgH2、TiFeH2、LaNi5H6)[23-25]等。盡管化學(xué)氫化物和絡(luò)合氫化物普遍具有較高的儲(chǔ)氫性能,但較高的工作溫度和較差的循環(huán)可逆性限制了它們的大規(guī)模應(yīng)用,此外,它們的合成和再生也是亟待解決的問題[26]。傳統(tǒng)金屬基氫化物(TiFeH2和LaNi5H6)可以在相對溫和的操作條件下可逆地吸氫與脫氫,但由于金屬含量較高,它們的質(zhì)量儲(chǔ)氫密度并不理想,例如,LaNi5的儲(chǔ)氫量只有1.4%(質(zhì)量分?jǐn)?shù))H2。因此,它們都不是固態(tài)儲(chǔ)氫材料最為合適的選擇。相比之下,MgH2的儲(chǔ)氫性能優(yōu)異(7.6%,質(zhì)量分?jǐn)?shù))、無毒無害、可逆性好,而且具備氫凈化功能,是一種極具前景的固態(tài)儲(chǔ)氫材料[27-28]。但是,其較高的脫氫焓(ΔH=76 kJ/mol)以及脫氫活化能(Ea=160 kJ/mol)導(dǎo)致MgH2具有較高的脫氫溫度[29-31]。此外,由于Mg—H鍵鍵能較高,H2分子的解離能力和H原子的重組能力較差,H原子在MgH2中擴(kuò)散系數(shù)較低等問題的存在,設(shè)計(jì)一種能在適宜溫度下快速實(shí)現(xiàn)吸脫氫的儲(chǔ)氫材料仍然是一個(gè)挑戰(zhàn)[32]。
本文首先介紹了MgH2的晶體結(jié)構(gòu)和儲(chǔ)氫機(jī)理,之后闡述了一系列改性方法,分析了不同改性方法所合成的MgH2儲(chǔ)氫復(fù)合材料的性能特點(diǎn),對其可逆吸脫氫量、熱力學(xué)和動(dòng)力學(xué)等性能進(jìn)行了探討。最后針對目前鎂基固態(tài)儲(chǔ)氫材料的研究進(jìn)展以及未來的挑戰(zhàn)進(jìn)行了簡要的總結(jié)與展望。
1 MgH2的晶格結(jié)構(gòu)和儲(chǔ)氫機(jī)理
為了改善MgH2的儲(chǔ)氫性能,必須全面了解其結(jié)構(gòu)穩(wěn)定性。MgH2在不同的溫度和壓力條件下會(huì)以不同的晶體結(jié)構(gòu)存在,分別是α-MgH2、β-MgH2、 γ -MgH2、 δ′ -MgH2、 ε -MgH2和cubic-MgH2。常見晶體結(jié)構(gòu)及相應(yīng)的壓力-溫度圖(P-T圖)如圖1和圖2所示[33]。已有研究證明MgH2的最低能態(tài)結(jié)構(gòu)是具有四方晶系金紅石型結(jié)構(gòu)的α-MgH2[34-35]。MgH2的P-T圖(圖2)表明,α-MgH2會(huì)在2 GPa 的壓力下轉(zhuǎn)變?yōu)棣?MgH2。隨著壓力由2 GPa 進(jìn)一步增加至7 GPa,MgH2的晶格結(jié)構(gòu)會(huì)實(shí)現(xiàn)由γ-MgH2向ε-MgH2的轉(zhuǎn)化。有報(bào)道稱,通過高壓扭轉(zhuǎn)方法增加應(yīng)變,α-MgH2可以完全轉(zhuǎn)變?yōu)棣?MgH2,具有離子鍵的γ 相氫鍵結(jié)合能力較弱,可使脫氫溫度降低80 K[36]。此外,在特定的壓力和溫度條件下,β-γ 和δ′-ε 有可能實(shí)現(xiàn)共存,不同結(jié)構(gòu)類型的MgH2共存可能會(huì)對氫解吸性能產(chǎn)生催化作用。例如在γ-MgH2和β-MgH2的共存體系中,前者會(huì)誘導(dǎo)后者的Mg—H鍵失穩(wěn)并釋放氫氣[37]。由于在相應(yīng)溫度和壓力范圍內(nèi),cubic-MgH2的吉布斯自由能高于其他五個(gè)相,因此cubic-MgH2沒有出現(xiàn)在P-T圖中。一系列現(xiàn)象證實(shí)了MgH2的吸脫氫性能與其晶體結(jié)構(gòu)具有密切關(guān)系。
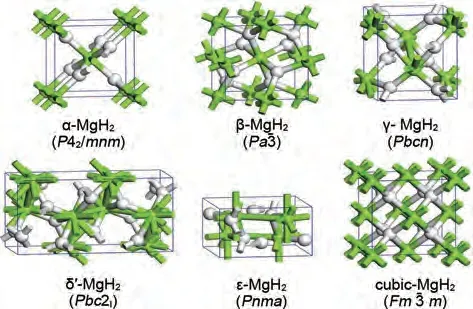
圖1 MgH2的常見晶體結(jié)構(gòu)[33]Fig.1 Common crystal structures of magnesium hydride [33]
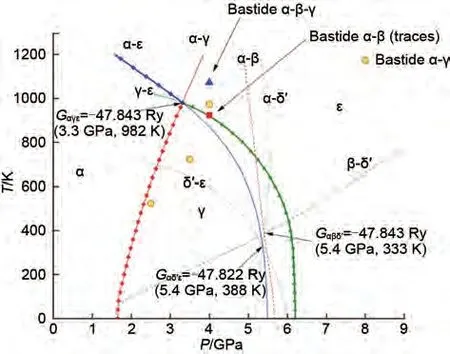
圖2 MgH2的P-T圖[33]Fig.2 The corresponding P-Tphase diagram of MgH2[33]
在闡述各種改性方法前需要對Mg/MgH2體系的儲(chǔ)氫機(jī)理進(jìn)行深入了解。Mg 在300~400 ℃,2.4~4 MPa條件下會(huì)與H2發(fā)生可逆化學(xué)反應(yīng)從而實(shí)現(xiàn)吸脫氫,其反應(yīng)方程式如式(1)所示:
正向?yàn)槲鼩浞艧岱磻?yīng),逆向?yàn)槊摎湮鼰岱磻?yīng)。如圖3(a)所示,MgH2的吸氫反應(yīng)由以下幾個(gè)步驟組成[38]:①物理吸附,即鎂通過范德華力將氫分子吸附至表面;②化學(xué)吸附,即解離的表面氫原子與鎂形成Mg—H鍵;③固溶體α相的形成,即氫擴(kuò)散到鎂的晶胞空隙并發(fā)生占據(jù);④固溶體β 相的形成,即晶胞中氫原子達(dá)到臨界濃度,產(chǎn)生新的穩(wěn)定相,PCT(壓力-組成-溫度)圖上會(huì)出現(xiàn)一個(gè)平坦的平臺,平臺寬度代表著可逆儲(chǔ)氫量;⑤β相生長和α 相的消失,平臺逐漸消失,實(shí)現(xiàn)固溶體α 相到β相的完全轉(zhuǎn)化。放氫反應(yīng)是吸氫反應(yīng)的逆過程。
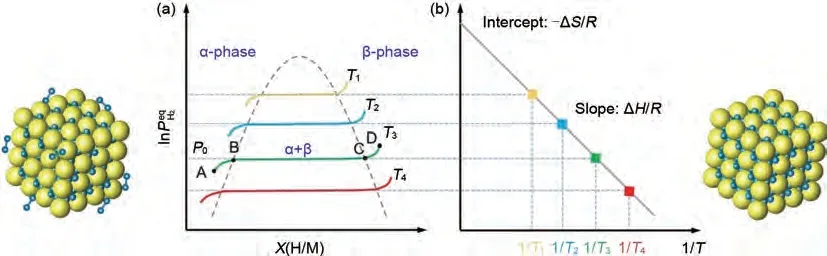
圖3 (a) Mg/MgH2的壓力成分等溫線圖[38];(b) Mg/MgH2相變相應(yīng)的范特霍夫圖[38]Fig.3 (a)Pressure component isotherm diagram of Mg/MgH2 and (b)Van′ t Hoff diagram corresponding to the Mg/MgH2 phase transition [38]
此外,通過測定不同溫度下的PCT 曲線,氫化鎂的脫氫焓(ΔH)和脫氫熵(ΔS)可通過Van't Hoff方程[圖3(b)]進(jìn)行計(jì)算,方程如式(2)所示:
其中,P0為大氣壓力(1.01×105Pa),ΔH和ΔS分別為MgH2的吸脫氫過程的焓變與熵變,T為吸放氫溫度[圖3(b)]。
2 MgH2的改性研究進(jìn)展
2.1 合金化
合金化是一種改善鎂基儲(chǔ)氫材料吸脫氫性能的常用方法。常見合金儲(chǔ)氫材料可分為稀土系(AB5型)、Zr系(AB2型)、Ti系(AB型)、鎂系(A2B型)。儲(chǔ)氫合金由兩部分組成,一部分為吸氫元素或與氫有很強(qiáng)親和力的元素A,它控制著儲(chǔ)氫量的多少,是組成儲(chǔ)氫合金的關(guān)鍵元素,主要是ⅠA~ⅤB 族金屬,如Ti、Zr、Ca、Mg、V、Nb、稀土元素Re等;另一部分則為吸氫量較小或根本不吸氫的元素B,它則控制著吸/脫氫的可逆性,如Fe、Co、Ni、Cr、Cu、Al等[39-41]。而各體系儲(chǔ)氫合金中,鎂系合金的儲(chǔ)氫質(zhì)量密度最高,被學(xué)術(shù)界一致認(rèn)為是目前最具應(yīng)用潛力的儲(chǔ)氫材料之一。典型的鎂基合金體系是通過加入過渡金屬元素或稀土元素與金屬鎂形成相應(yīng)的合金,其中過渡金屬元素可促進(jìn)H2分解為H原子,從而降低氫解離和重組的Ea。此外,過渡金屬未完全填充的空d電子軌道與氫原子相互作用會(huì)削弱Mg—H 鍵,從而降低儲(chǔ)氫材料的脫氫Ea與ΔH,實(shí)現(xiàn)快速脫氫[42-45]。而稀土元素可形成分散或包覆Mg/MgH2的稀土氫化物抑制循環(huán)過程中出現(xiàn)的團(tuán)聚現(xiàn)象,從而顯著改善鎂合金儲(chǔ)氫材料的循環(huán)穩(wěn)定性[46]。
鎂基二元固態(tài)合金儲(chǔ)氫材料的研究始于1967年,美國布魯克-海文國家實(shí)驗(yàn)室的Reilly等[47]首次研究Mg-Cu 系儲(chǔ)氫合金,但其僅有2.6%(質(zhì)量分?jǐn)?shù))的理論儲(chǔ)氫量,且Mg2Cu 和H2反應(yīng)形成的MgCu2不參與吸氫。為了改善儲(chǔ)氫性能,1968年,Reilly 等[48]通過混合熔煉首次制備了Mg2Ni 儲(chǔ)氫合金,該合金理論儲(chǔ)氫量提高至3.6%(質(zhì)量分?jǐn)?shù))。此外,Mg2Ni合金還具有較好的熱力學(xué)性能,Ni原子對H 的作用力強(qiáng)于Mg 原子對H 的作用,從而削弱了Mg—H鍵的相互作用。生成的Mg2NiH4二元?dú)浠锏拿摎洇降至64 kJ/mol,相比MgH2的76 kJ/mol 顯著降低,理論脫氫溫度也由MgH2的300~400 ℃高溫區(qū)間降至300 ℃以下。
鑒于Mg2NiH4合金優(yōu)異的儲(chǔ)氫性能,日本東北大學(xué)八木研究室[49]于1997 年首次采用氫化燃燒合成法(hydriding combustion synthesis,簡稱HCS法)直接合成了Mg2NiH4,該法合成過程在爐溫低于870 K條件下進(jìn)行,避免了鎂的揮發(fā),可直接從鎂鎳混合粉末制備出符合化學(xué)計(jì)量的鎂鎳儲(chǔ)氫合金。由于反應(yīng)中間產(chǎn)物Mg2Ni組織疏松、比表面積大和活性高的特點(diǎn),一步法即可直接獲得儲(chǔ)氫合金氫化物[50]。HCS法對Mg-Ni儲(chǔ)氫合金的制備方法進(jìn)行了創(chuàng)新,也是對Reilly 等[47-48]的研究進(jìn)行了補(bǔ)充與擴(kuò)展,為Mg2NiH4儲(chǔ)氫體系后續(xù)的快速發(fā)展與實(shí)際應(yīng)用奠定了基礎(chǔ)。
作為最具代表性的鎂基儲(chǔ)氫合金之一,Mg-Ni合金近年來也得到了更為深入的研究。Shao 等[51]機(jī)械球磨制備了Mg100-xNix系列合金,研究發(fā)現(xiàn),Mg50Ni50合金表現(xiàn)出最佳的吸氫動(dòng)力學(xué)和吸氫速率。該合金可以在373 K 下7 MPa 氫壓下吸附1.85%(質(zhì)量分?jǐn)?shù))H2(圖4)。Tan等[52]在Shao等[51]的研究基礎(chǔ)上,結(jié)合HSC 和機(jī)械球磨法合成了Mg100-xNix合金。差示掃描量熱(DSC,圖5)表明所有樣品均顯示兩個(gè)脫氫峰,分別對應(yīng)于β-MgH2和γ-MgH2。相比純Mg,Mg100-xNix合金的峰值脫氫溫度都有一定的下降。當(dāng)Ni 添加量為20 %時(shí),MgH2的初始/峰值脫氫溫度從320.3 ℃/340.4 ℃降低到223.9 ℃/247.3 ℃。表明Ni 對解吸溫度的降低有顯著的作用。此外,473 K 下Mg100-xNix合金均表現(xiàn)出快速的氫化動(dòng)力學(xué),在100 s 內(nèi)即可完成大于4%(質(zhì)量分?jǐn)?shù))的飽和吸氫量。
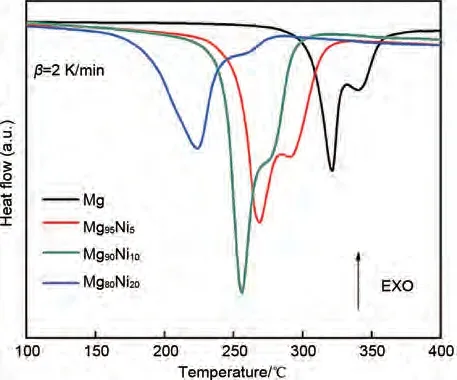
圖5 HCS+MM后Mg100-xNix(x=0、5、10和20)合金的DSC曲線[52]Fig.5 DSC curves of Mg100-xNix(x=0, 5, 10 and 20)alloys after HCS+MM [52]
眾所周知,空氣中的H2O和O2容易與主要儲(chǔ)氫元素Mg 反應(yīng)從而降低鎂合金的儲(chǔ)氫性能。為了解決這一難題,朱云峰團(tuán)隊(duì)[53]將HCS 法制備的Mg80Ni20Hx暴露在空氣中4個(gè)月后,研究了外部不利條件對Mg-Ni合金儲(chǔ)氫性能的影響。研究發(fā)現(xiàn),合成的Mg80Ni20Hx的脫氫峰值溫度為387.77 ℃,并且隨著空氣暴露時(shí)間的延長而顯著降低。在空氣中暴露4 個(gè)月后,脫氫峰溫度僅為239.83 ℃[圖6(a)]。TPD 圖[圖6(b)]顯示初始脫氫溫度從344 ℃下降到215 ℃。圖6(c)和(d)顯示了不同溫度下Mg80Ni20Hx在空氣中暴露4個(gè)月后的等溫吸/脫氫曲線。吸氫方面,在230 ℃、245 ℃、275 ℃和300 ℃下,接觸空氣后的儲(chǔ)氫合金在400 s內(nèi)吸收1.18%、2.59%、3.00%和3.04%(質(zhì)量分?jǐn)?shù))H2。在300 ℃下,空氣暴露后的Mg80Ni20Hx只需500 s即可實(shí)現(xiàn)飽和吸氫,遠(yuǎn)快于未接觸空氣的Mg80Ni20Hx(1500 s)。脫氫方面,在300 ℃下,接觸空氣后的儲(chǔ)氫合金在400 s內(nèi)脫附2.81%(質(zhì)量分?jǐn)?shù))H2。在230 ℃、245 ℃和275 ℃下,分別在2500 s內(nèi)脫附2.69%、2.80%和3.11%(質(zhì)量分?jǐn)?shù))H2。在300 ℃下,空氣暴露后的Mg80Ni20Hx可以在1000 s 內(nèi)完全脫氫,遠(yuǎn)快于未接觸空氣的Mg80Ni20Hx(2500 s)。 經(jīng)理論計(jì)算,Mg80Ni20Hx在空氣中暴露4個(gè)月后,其脫氫活化能Ea為(63.56±2.43) kJ/mol,較未接觸空氣的Mg80Ni20Hx(93.5 kJ/mol[54])明顯降低[圖6(e)、(f)]。
除了典型的Mg-Ni儲(chǔ)氫合金外,Al、Fe、Si以及鑭系元素也可與Mg 形成穩(wěn)定的Mg-Al 和Mg-稀土等二元合金相,在熱力學(xué)上大幅降低Mg 基材料的脫氫焓。不同Mg 基儲(chǔ)氫二元合金的儲(chǔ)氫容量及脫氫焓如表2所示。
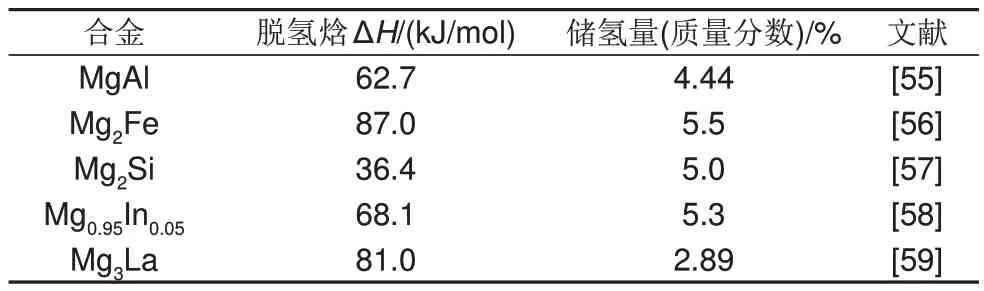
表2 一些二元鎂合金的儲(chǔ)氫量和脫氫焓Table 2 Hydrogen storage capacity and dehydrogenation enthalpy of some binary magnesium alloys
顯然,一些金屬元素的添加顯著改善了鎂基儲(chǔ)氫合金的動(dòng)力學(xué)性能和熱力學(xué)性能,但是形成的二元鎂基儲(chǔ)氫合金的儲(chǔ)氫量相較MgH2(7.6%,質(zhì)量分?jǐn)?shù))顯著降低。而當(dāng)過渡元素、Al 等非過渡金屬元素以及稀土元素復(fù)合添加時(shí),得到的鎂基復(fù)合材料的催化性能優(yōu)于只添加其中一種,儲(chǔ)氫量也有所提高。因此,為了兼顧高儲(chǔ)氫量和優(yōu)異的熱力學(xué)/動(dòng)力學(xué)性能,國內(nèi)外學(xué)者對鎂基儲(chǔ)氫多元合金體系進(jìn)行了深入探索,即向鎂中同時(shí)添加兩種或多種微量合金元素形成三元或高熵鎂合金。
Wang 等[60]通過煅燒制備了Mg-Al 合金,并通過機(jī)械球磨法將過渡金屬(TM=Ti、V、Ni)引入Mg-Al 合金中成功制備了Mg-Al-TM 三元合金。如圖7(a)所示,Mg-Al合金的起始吸氫溫度為188 ℃,在360 ℃時(shí)可逆儲(chǔ)氫容量達(dá)到4.21%(質(zhì)量分?jǐn)?shù)),而Mg-Al-TM(TM=Ti、V、Ni)三元合金的起始吸氫溫度為96 ℃,在360℃下的可逆儲(chǔ)氫容量(質(zhì)量分?jǐn)?shù))分別為4.28%、5.58%和5.77%。脫氫曲線[圖7(b)]表明,當(dāng)添加Ti、Ni和V后,Mg-Al合金的起始脫氫溫度(310 ℃)顯著降低。尤其是Mg-Al-V,其起始脫氫溫度降至244 ℃(比Mg-Al 合金低66 ℃)。Mg-Al-V合金具有最可觀的吸脫氫性能。
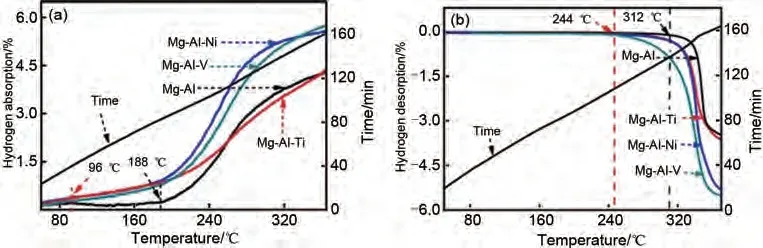
圖7 Mg-Al和Mg-Al-TM(TM=Ti, Ni, V)合金的吸脫氫曲線圖[60]Fig.7 Hydrogenation and dehydrogenation curves of the Mg-Al and Mg-Al-TM(TM=Ti、Ni、V)alloys [60]
Wei 等[61]通過真空感應(yīng)熔煉法成功制備了Mg95-xAl5Yx(x=0~5)合金。通過線性擬合Arrhenius 方程發(fā)現(xiàn):隨著Y 含量由x=0 增加到5,該鎂合金儲(chǔ)氫材料的脫氫活化能從(155.562±2.29) kJ/mol 下降至(79.622±5.61) kJ/mol(圖8),脫氫動(dòng)力學(xué)性能得到顯著改善。Mg-Al 基儲(chǔ)氫合金的初始脫氫溫度也隨著Y 含量的增加降低了40.2 ℃,最大儲(chǔ)氫量達(dá)到了5.1%(質(zhì)量分?jǐn)?shù)),顯著高于Mg-Al 二元合金(4.44%,質(zhì)量分?jǐn)?shù)),兼顧了高儲(chǔ)氫量和優(yōu)異的熱力學(xué)/動(dòng)力學(xué)性能。該三元合金儲(chǔ)氫性能的改善可歸因于合金中不同元素之間產(chǎn)生的YH3催化相和成核活性位點(diǎn)。
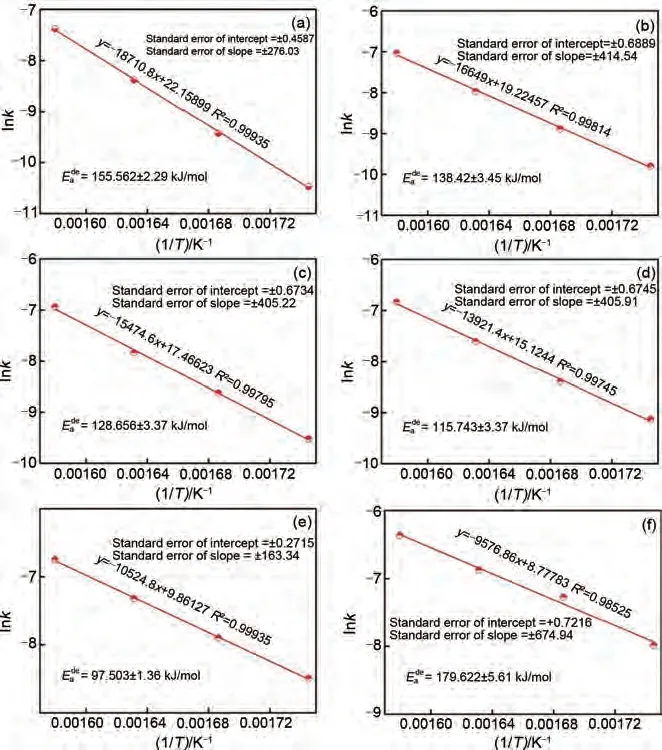
圖8 Mg95-xAl5Yx(x=0~5)的阿侖尼烏斯方程[61]:(a)Y0 alloys; (b)Y1 alloys; (c)Y2 alloys; (d)Y3 alloys; (e)Y4 alloys; (f)Y5 alloysFig.8 Arrhenius plots of the Mg95-xAl5Yx(x=0~5)[61]: (a)Y0 alloys; (b)Y1 alloys; (c)Y2 alloys; (d)Y3 alloys; (e)Y4 alloys and (f)Y5 alloys
針對稀土鎂合金儲(chǔ)氫體系,目前研究最多的是Mg-Ni-稀土三元合金。前文提到了典型的Mg-Ni儲(chǔ)氫二元合金的一些研究成果,而隨著儀器設(shè)備和相關(guān)理論的完善,Mg-Ni合金的儲(chǔ)氫溫度也由300 ℃以下進(jìn)一步降至250 ℃[62]。然而,Mg-Ni 二元合金的儲(chǔ)氫性能依舊不夠理想,仍有待進(jìn)一步提高。針對以上問題,相關(guān)學(xué)者通過將部分稀土元素引入其中成功制備了一系列Mg-Ni-稀土三元合金體系,為高性能Mg-Ni系儲(chǔ)氫合金成分設(shè)計(jì)與開發(fā)提供指導(dǎo)。
Yu 等[63]采用真空感應(yīng)爐法制備Mg95-x-Nix-Y5(x=5, 10, 15)三元合金。通過對鎳的組成、材料微觀形態(tài)、儲(chǔ)氫性能等進(jìn)行系統(tǒng)研究后發(fā)現(xiàn),YH3相在脫氫過程中不分解,均勻分散在母相合金中,對Mg/MgH2和Mg2Ni/Mg2NiH4的可逆相變起到了一定的催化和細(xì)化作用。通過密度泛函理論計(jì)算(density functional theory, DFT)發(fā)現(xiàn),Ni 元素的加入可以有效降低MgH2的能帶間隙,從而有效地改善鎂基儲(chǔ)氫合金的熱力學(xué)性能[64],Ni5、Ni10和Ni15樣品的脫氫焓分別降至84.5 kJ/mol、69.1 kJ/mol 和63.5 kJ/mol,Mg80Ni15Y5合金具有最佳的熱力學(xué)性能,該合金初始吸氫溫度也降至200 ℃。而吸脫氫過程中形成的YH3具有氫泵效應(yīng),使該三元儲(chǔ)氫合金材料能夠在1 min 內(nèi)吸附約5.4%(質(zhì)量分?jǐn)?shù))H2,達(dá)到了飽和儲(chǔ)氫容量的90 %,顯著改善了材料儲(chǔ)氫性能(圖9)。
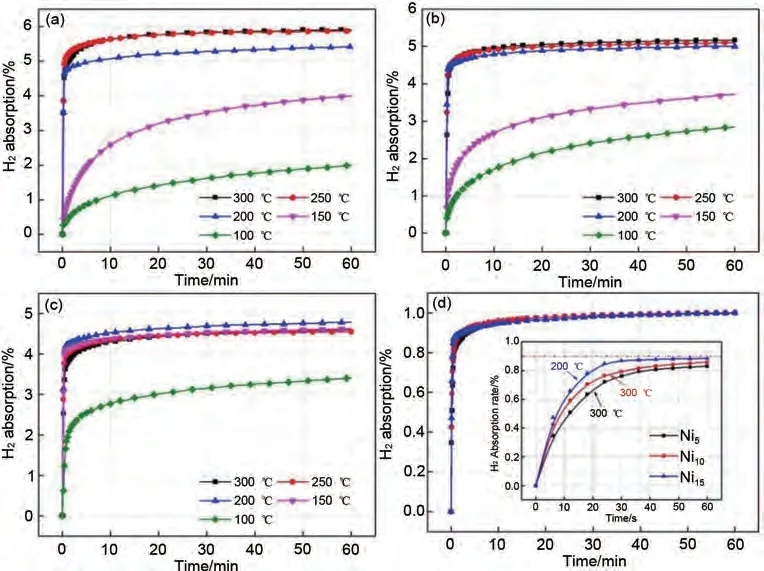
圖9 活化后的Mg95-XNixY5(x=5,10,15)樣品在不同溫度下的等溫吸氫曲線[63]:(a)Mg90Ni5Y5;(b)Mg85Ni10Y5;(c)Mg80Ni15Y5;(d)最佳氫化溫度下各樣品吸氫速率對比圖Fig.9 Isothermal hydrogen absorption curves of the activated Mg95-xNixY5(x=5, 10, 15)samples in different temperatures: (a)Mg90Ni5Y5; (b)Mg85Ni10Y5; (c)Mg80Ni15Y5 and (d)the comparison of hydrogen absorption rate of these samples at optimum hydrogenated temperature [63]
為了了解鎂基儲(chǔ)氫合金的長期循環(huán)機(jī)理,Guo等[65]研究了Mg-Ni-La三元合金在623 K條件下經(jīng)過200次循環(huán)后的循環(huán)可逆性。研究發(fā)現(xiàn),Mg-Ni-La合金具有較好的脫氫循環(huán)性能,200次循環(huán)后脫氫量仍能保持在5.5%(質(zhì)量分?jǐn)?shù))以上(圖10)。隨著循環(huán)次數(shù)的增加,樣品的脫氫速率先增大后減小,最終達(dá)到相對穩(wěn)定的狀態(tài)。微觀結(jié)構(gòu)觀察發(fā)現(xiàn),在吸/脫氫過程中,由于體積的反復(fù)膨脹和收縮,納米合金粉末同時(shí)存在燒結(jié)和氫爆兩種現(xiàn)象。同時(shí),原位形成的LaHx(x=2, 3)納米晶體可在一定程度上抑制粉末的燒結(jié)。經(jīng)過200次循環(huán)后,實(shí)驗(yàn)樣品的平均粒徑減小,比表面積明顯增大,導(dǎo)致MgH2和Mg2NiH4的分解溫度輕微向低溫轉(zhuǎn)移。此外,Mg2NiH4和LaHx(x=2, 3)在長期循環(huán)過程中被證明是穩(wěn)定的催化劑,經(jīng)過200次循環(huán)后仍能均勻分布在粉末中。
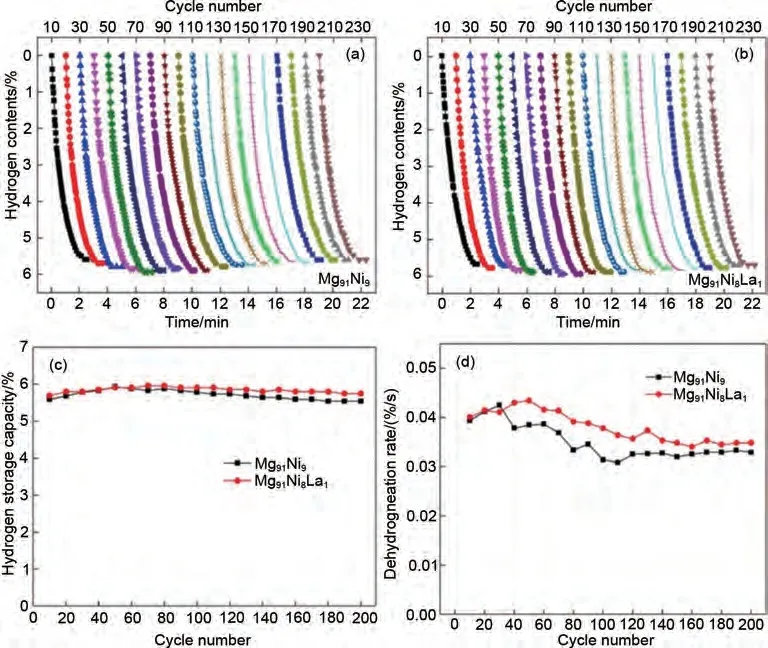
圖10 Mg91Ni9與Mg91Ni8La1的200次循環(huán)的脫氫性能圖[65]Fig.10 200-cycle dehydrogenation performance diagram of Mg91Ni9 and Mg91Ni8La1[65]
可以看出,三元儲(chǔ)氫合金展現(xiàn)出更加優(yōu)異的熱力學(xué)與動(dòng)力學(xué)性能,儲(chǔ)氫量也有了明顯的提高,近年來報(bào)道的一些Mg 基三元與四元合金相關(guān)儲(chǔ)氫參數(shù)如表3所示。
表3 一些鎂合金的相應(yīng)儲(chǔ)氫參數(shù)Table 3 Corresponding hydrogen storage parameters of some magnesium alloys
雖然前文提及的一系列三元鎂合金展現(xiàn)出了比簡單二元合金更加理想的儲(chǔ)氫性能,但是總體來說,三元鎂合金的脫氫溫度和脫氫活化能等參數(shù)需要進(jìn)一步改善。由此一種新型合金——高熵合金(HEA)進(jìn)入了學(xué)者們的研究視野中。
高熵合金由中國臺灣學(xué)者葉筠蔚等[74]于1995年首次提出,打破了傳統(tǒng)合金固有的設(shè)計(jì)思路。高熵合金是指由不少于5種元素所組成的合金,各原子占比最大不超過35%最小不低于5%,而傳統(tǒng)合金則含有一種原子百分占比大于50%的元素。近年來,將高熵鎂合金作為儲(chǔ)氫合金進(jìn)行研究的相關(guān)報(bào)道還很少,但由于多個(gè)不同半徑原子之間理化性質(zhì)差異導(dǎo)致的嚴(yán)重晶格畸變?yōu)闅湓映珊撕蛿U(kuò)散提供了更多的空間,這使得高熵鎂合金成為了極具潛力的儲(chǔ)氫合金。各國學(xué)者也開始對高熵儲(chǔ)氫鎂合金進(jìn)行了深入的探索。
申炳澤等[75]通過機(jī)械球磨法制備了MgxTiVNiAlCr(x=0, 0.5, 1)。圖11 為高熵合金TiVNiAlCr、Mg0.5TiVNiAlCr 以及MgTiVNiAlCr 分別在473 K、523 K 以及573 K 下的等溫吸氫曲線。可以看出,各試樣隨著溫度的升高吸氫量都會(huì)有一定程度的下降,因?yàn)榇蠖鄶?shù)吸附都是放熱過程,溫度升高吸附量會(huì)降低,所以簡單的升溫來促進(jìn)氫氣的熱擴(kuò)散是不可取的。在473 K 溫度條件下TiVNiAlCr、Mg0.5TiVNiAlCr 以及MgTiVNiAlCr 的最大儲(chǔ)氫量(質(zhì)量分?jǐn)?shù))分別為0.57%、0.7%、1.3%。儲(chǔ)氫量隨著Mg 含量的增加而顯著改善,但是儲(chǔ)氫量較低。總的來說,Mg 元素的添加對高熵合金的儲(chǔ)氫性能有所提高,但仍無法達(dá)到理想的儲(chǔ)氫量,有待進(jìn)一步改善。
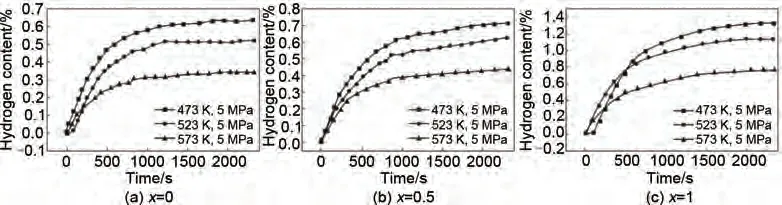
圖11 高熵合金MgxTiVNiAlCr在473 K、523 K、573 K下的等溫吸氫曲線[75]Fig.11 Hydrogen absorption curves of MgxTiVNiAlCr(x=0, 0.5, 1)at 473 K, 523 K, 573 K [75]
為了進(jìn)一步提高含鎂高熵合金的儲(chǔ)氫量,Montero 等[76]在惰性氣氛下,采用機(jī)械化學(xué)合成方法制備了一種新型高熵合金Mg0.1Ti0.3V0.25Zr0.1Nb0.25。該合金在25 ℃室溫下迅速吸氫,在1 分鐘內(nèi)達(dá)到1.72H/M(H/M 指單位質(zhì)量材料的儲(chǔ)氫能量)的最大吸氫量,吸氫速率與吸氫量相比上文提到的MgTiVNiAlCr(1.3%,質(zhì)量分?jǐn)?shù))都有了明顯的改善。經(jīng)過一次吸脫氫循環(huán)后,Mg0.1Ti0.3V0.25Zr0.1Nb0.25合金的儲(chǔ)氫量有略微下降。吸脫氫循環(huán)性能測試(圖12)表明Mg0.1Ti0.3V0.25Zr0.1Nb0.25五元合金在第二個(gè)循環(huán)中儲(chǔ)氫容量損失了約11%(質(zhì)量分?jǐn)?shù)從2.7%降到2.41%),隨后幾個(gè)循環(huán)的可逆容量穩(wěn)定在2.4%左右。這與未添加鎂元素的Ti0.325V0.275Zr0.125Nb0.275四元合金形成鮮明對比,Ti0.325V0.275Zr0.125Nb0.275在前4 次循環(huán)中損失近28%的儲(chǔ)氫容量,然后達(dá)到約1.9%(質(zhì)量分?jǐn)?shù))的穩(wěn)定值。顯然,含Mg 的HEA 比四元HEA(不含Mg)具有更大的可逆儲(chǔ)氫量。
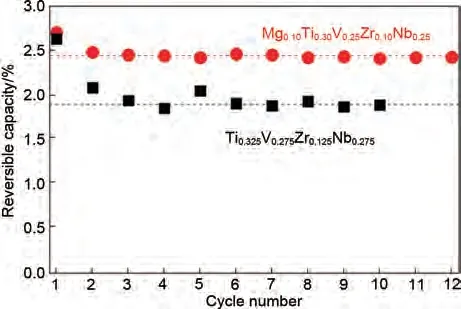
圖12 Mg0.1Ti0.3V0.25Zr0.1Nb0.25和Ti0.325V0.275Zr0.125Nb0.275合金循環(huán)過程中可逆儲(chǔ)氫容量對比圖[76]Fig.12 Comparison between the variation of the reversible hydrogen storage capacity during cycling for the quinary Mg0.1Ti0.3V0.25Zr0.1Nb0.25 and quaternary Ti0.325V0.275Zr0.125Nb0.275 alloys [76]
本節(jié)具體概述了鎂基二元、三元、四元以及高熵儲(chǔ)氫合金各自的優(yōu)缺點(diǎn)和發(fā)展脈絡(luò)。總的來說,合金化可以有效降低脫氫溫度以及脫氫焓變等參數(shù),同時(shí)兼顧穩(wěn)定的循環(huán)吸脫氫性能。但是在合金化過程中由于引入大量金屬元素導(dǎo)致了儲(chǔ)氫合金氫容量的大幅下降。
2.2 納米化
納米化是同時(shí)調(diào)節(jié)MgH2熱力學(xué)和動(dòng)力學(xué)的改性策略。由于納米材料的量子尺寸效應(yīng)、小尺寸效應(yīng)和表面效應(yīng),與大體積MgH2相比,納米MgH2具有更高的氫擴(kuò)散系數(shù)以及良好的吸/脫氫動(dòng)力學(xué)。理論計(jì)算表明,當(dāng)晶粒尺寸在11~1.3 nm時(shí),氫解吸活化能顯著降低。當(dāng)粒徑降至0.9 nm 時(shí),解吸溫度下降到200 ℃[77-79]。已報(bào)道的制備納米鎂基材料的納米技術(shù)包括高能球磨法[80]、化學(xué)還原法[81]、氣相沉積法[82]、納米限域法[83]等。
2.2.1 高能球磨法
高能球磨(HEBM)是制備鎂納米顆粒廣泛使用的方法之一,球磨過程形成了不穩(wěn)定的γ-MgH2相、應(yīng)力、應(yīng)變、晶體缺陷以及大量的納米晶界和相界,可在一定程度上改善MgH2的儲(chǔ)氫性能。Schulz等[84]首次發(fā)現(xiàn)高能球磨可以獲得具有高比表面積的新型納米結(jié)構(gòu),在300 ℃下400 s內(nèi)吸收7%(質(zhì)量分?jǐn)?shù))H2,在350 ℃下600 s內(nèi)脫附7%(質(zhì)量分?jǐn)?shù))H2。結(jié)果表明,機(jī)械球磨引起的結(jié)構(gòu)缺陷降低了吸/脫氫的活化能。Varin等[85]進(jìn)一步研究了MgH2的平均粒徑隨不同研磨時(shí)間的變化,發(fā)現(xiàn)當(dāng)粒徑減小到600 nm時(shí),脫氫溫度比純MgH2低60 ℃。
然而高能球磨法易引入雜質(zhì)顆粒,容易分布不均,并且易引起材料的團(tuán)聚和長大,使得循環(huán)穩(wěn)定性降低。
介質(zhì)阻擋放電等離子體輔助球磨(DBDP)是Ouyang 等[86]發(fā)展的一種改進(jìn)球磨技術(shù)。由于材料不僅受到機(jī)械球磨機(jī)的粉碎和沖擊,而且還受到高能非平衡等離子體的熱沖擊和活化效應(yīng),DBDP的研磨效率明顯高于HEBM。Dan等[87]通過等離子體球磨工藝在MgH2表面合成了超細(xì)鎳納米粒子(2~6 nm)。復(fù)合材料在275 ℃下10分鐘內(nèi)可快速脫附超過6.5%(質(zhì)量分?jǐn)?shù))H2,并且在九個(gè)吸/脫氫循環(huán)中幾乎沒有容量衰減(圖13)。
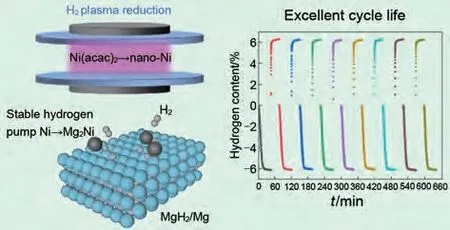
圖13 納米MgH2-Ni體系的催化機(jī)理圖及循環(huán)性能圖[87]Fig.13 Schematic of the catalytic mechanism and cycle properties of the nano-MgH2-Ni system [87]
DBDP為制備納米材料和提高球磨效率提供了一種可行、低成本、無污染的方法,對先進(jìn)鎂基儲(chǔ)氫材料的設(shè)計(jì)和制備具有重要的理論指導(dǎo)意義。
氫化燃燒合成(HCS)以其活性高、無活化產(chǎn)物以及產(chǎn)物吸氫速率快而著稱,相關(guān)科研人員結(jié)合HCS 與HEBM 進(jìn)行了廣泛的研究。Liu 等[88]結(jié)合氫化燃燒合成和高能研磨(HCS+HEBM)制備了納米結(jié)構(gòu)化的Mg2Ni,發(fā)現(xiàn)吸氫速率在313 K 和373 K明顯增加。初始脫氫溫度為370 K,比HCS產(chǎn)物低約190 K。最近,韓樹民團(tuán)隊(duì)[89]通過HCS+MM 將MgC0.5Co3化合物引入MgH2。材料在325 ℃條件下60 min 內(nèi)脫氫量達(dá)到4.38%(質(zhì)量分?jǐn)?shù)),接近純MgH2的4.5 倍。此外,解吸活化能從純MgH2的(162.8±8.3) kJ/mol 降 低 到(126.7±1.4) kJ/mol。MgC0.5Co3保持相對穩(wěn)定,循環(huán)后沒有發(fā)生任何化學(xué)轉(zhuǎn)化(圖14)。
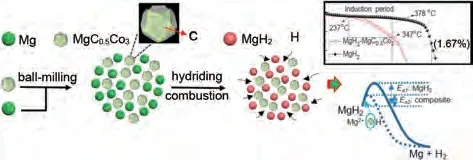
圖14 MgC0.5Co3催化機(jī)理圖[89]Fig.14 Diagram of the catalytic mechanism of MgC0.5Co3[89]
2.2.2 化學(xué)還原法
化學(xué)還原是合成Mg/MgH2納米顆粒的有效方法。Rieke等[90]于1972年首次通過堿金屬還原鎂鹽成功獲得了高活性鎂粉。之后,雙環(huán)戊二烯基鎂(MgCp2)、二正丁基鎂(MgBu2)和氯化鎂等也被選為Mg源得到了廣泛研究[91-93]。化學(xué)還原通常會(huì)引入電子載流子來加速電子在堿金屬和鎂鹽之間的轉(zhuǎn)移過程。由于鎂的低氧化還原電位(EMg2+/Mg=-2.37 V),鎂鹽在非水溶液的非質(zhì)子溶劑中被電化學(xué)還原,整個(gè)體系由作為電解質(zhì)的表面活性劑以及納米顆粒穩(wěn)定劑組成。2018 年,Shen 等[94]以TBAB 為表面活性劑,通過對Mg(BH4)2的電化學(xué)還原,獲得了Mg 20納米顆粒(60~100 nm)。Mg納米顆粒在100 ℃下氫化后,完全轉(zhuǎn)化為β-MgH2(70.4 %)和γ-MgH2(29.6 %)。γ-MgH2在一個(gè)循環(huán)后穩(wěn)定存在,γ-MgH2的存在使得脫氫活化能下降到(69.1±2.9) kJ/mol,脫氫焓下降至(57.5±5.3) kJ/mol。Jeon 等[95]報(bào)道了在含有鋰、萘和聚甲基丙烯酸甲酯(PMMA)的四氫呋喃溶液中還原二茂鎂(MgCp2)的方法,獲得了能隔絕水氧的聚合物基納米Mg 復(fù)合材料(Mg NCs/PMMA),Mg NCs 粒徑約為5 nm。在200 ℃下30 min內(nèi)可以快速吸附4%(質(zhì)量分?jǐn)?shù))H2,PMMA有效抑制了循環(huán)過程中Mg/MgH2的團(tuán)聚與粗化(圖15)。Francois 等[96]以溴化四丁胺(TBA)為模板在四氫呋喃中電化學(xué)還原制備了5 nm的Mg納米膠體,該材料在室溫下能夠可逆儲(chǔ)存7.6%(質(zhì)量分?jǐn)?shù))H2,在85 ℃下完全脫氫。Norberg等[97]在聯(lián)苯、苯或萘為電子載體的情況下,用鉀(K)還原MgCp2制備了25~38 nm的鎂單晶,發(fā)現(xiàn)25 nmMg納米晶體表現(xiàn)出最好的吸/脫氫性能。
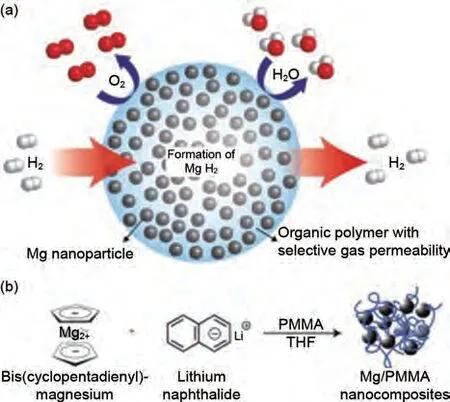
圖15 Mg NCs/PMMA在空氣暴露條件下的儲(chǔ)氫機(jī)理圖[95]Fig.15 Diagram of the mechanism of hydrogen storage under air exposure of Mg NCs/PMMA [95]
然而,化學(xué)還原法難以控制顆粒的規(guī)模和形態(tài),合成的鎂顆粒的形貌無明顯差異。此外,萘、聯(lián)苯或苯等電子載體會(huì)保留在儲(chǔ)氫材料中,一定程度上抑制儲(chǔ)氫性能。
2.2.3 氣相沉積法
除了高能球磨、化學(xué)還原法外,氣相沉積也是一種常見的納米改性方法。氣相沉積法是基于電弧產(chǎn)生高溫使金屬瞬間蒸發(fā),在氫氣等氣體作用下使金屬原子經(jīng)歷蒸發(fā)、形核、長大、凝聚等一系列過程,以獲得納米Mg。例如,Matsumoto 等[98]通過調(diào)節(jié)溫度和氫壓,利用化學(xué)氣相沉積(CVD)成功制備了三種形貌的MgH2。Li 等[99]通過物理氣相沉積(PVD)合成了直徑為30~50 nm、80~100 nm 和150~170 nm 的Mg 納米線,發(fā)現(xiàn)它們的脫氫活化能和脫氫焓變隨著納米線直徑的減小而減小。直徑最小(30 nm)的Mg 納米線在300 ℃下30 min 內(nèi)吸收7.6%H2并釋放6.8%H2。后來,該課題組[99]構(gòu)建了直徑更小的Mg 納米線理論模型,當(dāng)納米線直徑只有0.85 nm 時(shí),脫氫焓只有34.5 kJ/mol。但是,到目前為止,這種尺寸的納米線還難以通過實(shí)驗(yàn)合成。Lu 等[100]采用氣相沉積法制備了核殼狀的Mg@Pt納米顆粒(約200 nm)。Mg3Pt充當(dāng)MgH2放氫的“氫泵”,改善了吸脫氫動(dòng)力學(xué),初始脫氫溫度由MgH2的333.3 ℃降至287.5 ℃。但脫氫溫度仍然難以滿足實(shí)際應(yīng)用條件,有待進(jìn)一步改善。
此外,氣相沉積法還可用于制造多層納米級Mg/MgH2薄膜[101]。Higuchi 等[102]通過射頻磁控濺射設(shè)計(jì)了Pd(50 nm)/Mg(xnm)/Pd(50 nm)三明治結(jié)構(gòu)薄膜(圖16)。脫氫溫度降低至100 ℃以下。薄膜中的壓縮應(yīng)力和PdH2對MgH2的氫泵效應(yīng)導(dǎo)致了脫氫溫度的下降。

圖16 Pd (25 nm)/Mg (200 nm)(a)和Pd (50 nm)/Mg(200 nm)/Pd (50 nm)(b)薄膜截面的TEM圖[101]Fig.16 TEM micrographs for the cross section of (a)Pd (25 nm)/Mg (200 nm) and (b) Pd (50 nm)/Mg (200 nm)/Pd (50 nm) films [101]
多層膜結(jié)構(gòu)有利于研究相應(yīng)的催化機(jī)理。其組成、界面和結(jié)晶度可以在納米尺度上精確調(diào)整,但制備過程復(fù)雜且材料昂貴。需要進(jìn)一步開發(fā)適合實(shí)際生產(chǎn)條件的沉積方法。
2.2.4 納米限域法
納米限域(簡稱NC)是將Mg/MgH2納米團(tuán)簇限制在多孔支架中,多孔支架作為納米材料的團(tuán)聚抑制劑和尺寸控制劑顯著改善了鎂基材料的儲(chǔ)氫性能。石墨烯和碳納米管等碳材料作為支架已廣泛應(yīng)用于MgH2的改性研究[103-104]。Xia 等[105]在石墨烯的結(jié)構(gòu)導(dǎo)向效應(yīng)下,通過加氫反應(yīng)誘導(dǎo)自組裝制備了均勻分布的單分散MgH2納米顆粒,該材料在150 ℃下7 min內(nèi)可吸附5.4%(質(zhì)量分?jǐn)?shù))H2。MgH2與石墨烯之間的強(qiáng)耦合保證了納米結(jié)構(gòu)的穩(wěn)定性。此外,石墨烯的高熱導(dǎo)率可以促進(jìn)熱擴(kuò)散,提高氫氣的吸附/脫附速率,從而獲得了優(yōu)異的性能。除碳基材料外,聚甲基丙烯酸甲酯(PMMA)、金屬-有機(jī)框架(MOFs)以及介孔CoS納米盒(CoS-NB)作為支架材料也得到了廣泛研究。Ruminski 等[106]將不同比例的Mg納米顆粒包覆在PMMA中,研究了包覆聚合物對納米晶鎂的空氣穩(wěn)定性和儲(chǔ)氫密度的影響(圖17)。含65%(質(zhì)量分?jǐn)?shù))Mg-PMMA 復(fù)合材料在空氣中吸收了6.95%(質(zhì)量分?jǐn)?shù))H2,在空氣中暴露3 個(gè)月后幾乎沒有氧化,而含33.2%(質(zhì)量分?jǐn)?shù))Mg-PMMA復(fù)合材料僅吸收了4.86%(質(zhì)量分?jǐn)?shù))H2,在空氣中完全氧化。研究表明,聚合物用量的減少提高了Mg-PMMA復(fù)合材料的空氣穩(wěn)定性和吸氫能力。Lim 等[107]通過對MOF 中雙環(huán)戊二烯基鎂(MgCp2)的熱分解,制備了嵌在MOF 中的Mg 納米晶體。它具有物理吸附和化學(xué)吸附的雙重特性,表現(xiàn)出降低化學(xué)吸附/脫附溫度的協(xié)同效應(yīng)。鄒建新團(tuán)隊(duì)[108]通過氫化浸漬在Ni-MOF 中的MgBu2,制備了尺寸約為3 nm的MgH2顆粒,研究表明,MgH2@Ni-MOF復(fù)合材料中Mg/MgH2的熱力學(xué)[吸附/脫附焓分別為(65.7±2.1) kJ/mol 和(69.7±2.7) kJ/mol]和動(dòng)力學(xué)[吸附/脫附活化能分別為(41.5±3.7) kJ/mol 和(144.7±7.8) kJ/mol]均顯著改善。MgH2@Ni-MOF在熱力學(xué)和動(dòng)力學(xué)方面的改進(jìn)歸因于納米約束的Mg/MgH2的“納米效應(yīng)”和原位形成的Mg2Ni/Mg2NiH4的催化效應(yīng)。之后其團(tuán)隊(duì)[109]又通過模板法制備了限域在CoS-納米盒(CoS-NBs)支架中的MgH2@CoS-NBs。MgH2@CoS-NBs 復(fù) 合 材 料 在548 K下2小時(shí)內(nèi)脫附1.76%(質(zhì)量分?jǐn)?shù))H2。即使在較低的溫度(498 K)下,復(fù)合材料仍然可以在2小時(shí)內(nèi)脫附1.13%(質(zhì)量分?jǐn)?shù))H2。相比之下,純MgH2樣品在548 K 和498 K 下2 小時(shí)內(nèi)只能脫附0.42%和0.16%(質(zhì)量分?jǐn)?shù))H2。該復(fù)合材料經(jīng)過10次的吸/脫氫循環(huán)后,吸氫動(dòng)力學(xué)和吸氫能力方面沒有明顯的降低跡象,MgH2@CoS-NBs 復(fù)合材料優(yōu)異的吸氫動(dòng)力學(xué)和循環(huán)穩(wěn)定性主要?dú)w因于納米限制的Mg/MgH2晶體的“納米尺寸效應(yīng)”、MgS的催化作用和CoS-NBs支架的多功能作用。MgH2@CoS-NBs復(fù)合材料的具體制備過程與催化機(jī)理以及循環(huán)吸脫氫性能見圖18。毫無疑問,納米支架結(jié)構(gòu)通過將其顆粒尺寸限制在納米范圍內(nèi),縮短了氫氣的擴(kuò)散路徑,大大提高了MgH2的儲(chǔ)氫熱力學(xué)/動(dòng)力學(xué)性能。然而,目前尚不清楚所觀察到的增強(qiáng)效果是由于自身的限域性,還是由于氫化物與支架之間的化學(xué)相互作用、氫化物-溶劑加合物的形成等其他因素導(dǎo)致的。此外,體系儲(chǔ)氫能力差的問題阻礙了這種方法在大規(guī)模儲(chǔ)氫系統(tǒng)制備中的應(yīng)用。
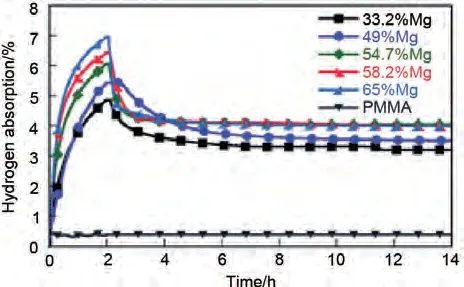
圖17 Mg-PMMA 納米復(fù)合材料的吸氫性能圖[106]Fig.17 Hydrogen absorption of Mg-PMMA nanocomposites containing 33.2%, 49%, 54.7%,58.2% and 65% Mg [106]
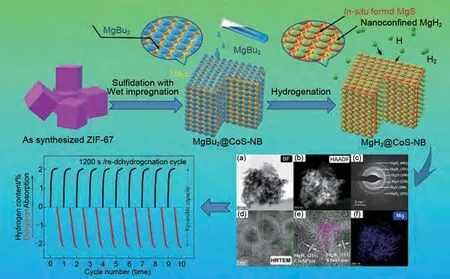
圖18 MgH2@CoS-NBs的制備過程、催化相表征和循環(huán)性能圖[109]Fig.18 Preparation process, catalytic phase characterization, and cycling performance diagram of MgH2@Co S-NBs [109]
本節(jié)就實(shí)現(xiàn)納米化的各種制備方法進(jìn)行了詳細(xì)介紹,為了更直觀地比較不同方法制備的納米Mg/MgH2儲(chǔ)氫性能方面的差異,表4對其進(jìn)行了總結(jié)。
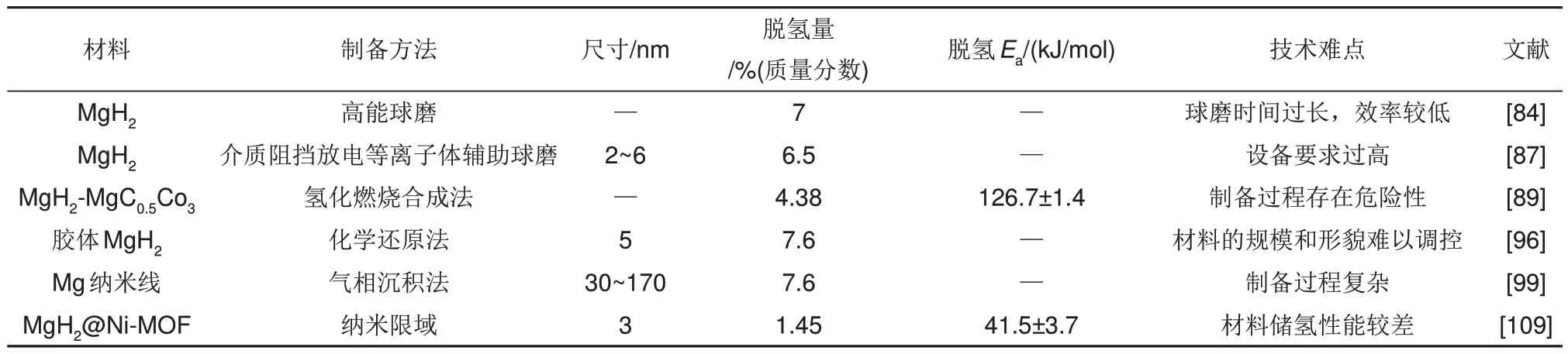
表4 不同方法制備的MgH2的性能差異和技術(shù)難點(diǎn)Table 4 Performance differences and technical difficulties of MgH2 prepared by different methods
2.3 催化劑改性
引入催化劑可以通過降低活化能,在一定程度上改善MgH2的吸/脫氫動(dòng)力學(xué)。在過去的幾年中,眾多催化劑得到了廣泛的探索和研究,包括金屬、金屬氧化物、其他金屬化合物、金屬與碳基復(fù)合催化劑、高熵合金等。本節(jié)將分別闡述不同種類的催化劑對鎂基材料儲(chǔ)氫性能的影響和作用機(jī)制。
2.3.1 過渡金屬改性
研究發(fā)現(xiàn)[110-111],過渡金屬往往有很多空軌道和占據(jù)軌道(軌道上有大量的d 電子),可以用來進(jìn)行電荷轉(zhuǎn)移、成鍵,因此過渡金屬對氫原子具有很強(qiáng)的親和力。在氫分子的解離或者氫原子的重組過程中,過渡金屬的d電子和氫原子/氫分子軌道上的電子發(fā)生轉(zhuǎn)移填充,由此產(chǎn)生的相互作用力可大幅降低氫分子解離或者氫原子重組的活化能,從而改善鎂基材料的吸脫氫性能。過渡金屬催化劑可分為Ti、Ni、Fe三大類。
針對過渡金屬Ti,在1999年Liang等[112]用機(jī)械球磨法制備了MgH2-TM(TM=Ti、F、Mn、Fe、Ni)納米復(fù)合材料。研究得出,含Ti的復(fù)合材料表現(xiàn)出最佳的解吸動(dòng)力學(xué),MgH2-5%(原子分?jǐn)?shù))Ti 復(fù)合材料在250 ℃下可在1000 s 內(nèi)完全脫氫。Pukazhselvan 等[113]深入探討了Ti 的催化機(jī)理,發(fā)現(xiàn)在Ti/MgH2納米復(fù)合材料中通過強(qiáng)機(jī)械球磨可以形成穩(wěn)定的TiH2-x。在脫/再氫化過程中,TiH2-x會(huì)轉(zhuǎn)化為TiH2,使MgH2的脫氫活化能降低到89.4 kJ/mol,微觀分析表明,TiH2晶體的尺寸約為Ti 晶體的4~5倍,表明催化劑引入引起的晶格應(yīng)變是改變MgH2儲(chǔ)氫性能的重要因素。
相比于簡單球磨制備的MgH2-Ti 儲(chǔ)氫材料,許多學(xué)者還使用一些新的制備方法將金屬Ti直接引入Mg 當(dāng)中,成功制備了具有優(yōu)異吸脫氫性能的儲(chǔ)氫材料。
Choi 等[114]采用化學(xué)蒸氣合成(CVS)法制備Mg-5%(質(zhì)量分?jǐn)?shù))Ti納米粉體混合物,該方法一定程度上抑制了材料的團(tuán)聚現(xiàn)象(圖19),Mg-Ti 粉末幾乎是球形的,直徑為50~500 nm,只有小部分顆粒發(fā)生了輕微的團(tuán)聚。圖20 熱重分析(TGA)表明CVS法制備的吸氫態(tài)MgH2-5%(質(zhì)量分?jǐn)?shù))Ti納米混合物(曲線A)的脫氫溫度降至190 ℃左右,分別比商用MgH2(曲線B)和Ti球磨引入的MgH2(曲線C)的起始溫度低約191 ℃和88 ℃。Mg-Ti 納米粉末在70 分鐘內(nèi)實(shí)現(xiàn)完全脫氫。而商用MgH2和Ti 球磨引入的MgH2需要124 分鐘以上。三個(gè)樣品的起始脫氫溫度和完全脫氫時(shí)間存在顯著差異,表明與商用MgH2和Ti球磨引入的MgH2相比,通過化學(xué)蒸氣合成(CVS)法制備的Mg-5%(質(zhì)量分?jǐn)?shù))Ti 納米材料的脫氫性能有了進(jìn)一步改善,也證明了探究不同制樣方法來改善材料性能的重要性。

圖19 鎂鈦納米粉末的TEM圖[114]Fig.19 TEM image of the Mg-Ti nanopowder [114]
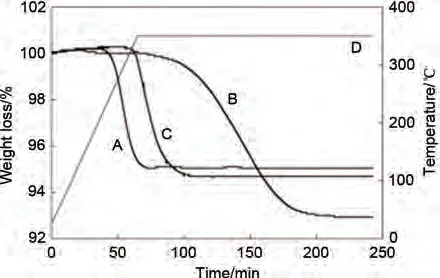
圖20 Mg-Ti納米粉末的氫化產(chǎn)物(A)MgH2-5%(質(zhì)量分?jǐn)?shù))Ti(CVS)、(B)商用MgH2、(C)MgH2-5%(質(zhì)量分?jǐn)?shù))Ti(球磨)的TGA曲線 [114]Fig.20 TGA profiles of the hydrogenated products of the Mg-Ti nanopowder(A)MgH2-5%Ti(CVS); (B)commerical MgH2; (C)MgH2-5%Ti(milled)[114]
針對過渡金屬Ni,早在2005 年Hanada 等[115]就通過球磨將商用MgH2粉末與金屬Ni混合,得到MgH2-Ni復(fù)合材料。如圖21所示,通過熱解吸質(zhì)譜(TDMS)發(fā)現(xiàn)球磨2 小時(shí)后的Ni 引入復(fù)合材料的脫氫溫度降至260 ℃,遠(yuǎn)低于純MgH2(370 ℃)。球磨1小時(shí)導(dǎo)致起始脫氫溫度降低至約250 ℃,而球磨15分鐘導(dǎo)致峰值溫度進(jìn)一步降低至約210℃。由于球磨時(shí)間過短導(dǎo)致MgH2與Ni混合不均勻,峰值信號較弱。但通過控制球磨時(shí)間,Ni展現(xiàn)出了優(yōu)異的催化效果。
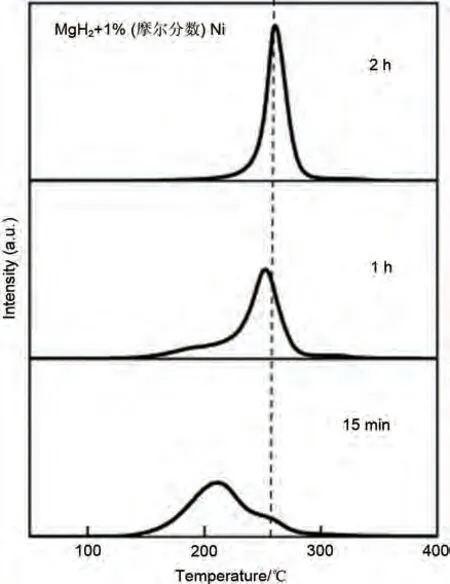
圖21 1 MPa氫氣氣氛400 r/min下研磨15 min、1 h、2 h的MgH2-1%(摩爾分?jǐn)?shù))Ni的熱解吸質(zhì)譜[115]Fig.21 Thermal desorption mass spectra of hydrogen from the MgH2 composite with 1 mol% Ni by milling for 15 min, 1 h, and 2 h at 400 r/min under a hydrogen gas atmosphere of 1 MPa [115]
Ni 納米粒子的優(yōu)異催化作用得到證實(shí)。近年來,催化劑用量和顆粒尺寸等其他因素也得到了廣泛研究。Xie等[116]研究了引入不同含量Ni納米顆粒的MgH2的儲(chǔ)氫動(dòng)力學(xué)。等溫脫氫曲線(見圖22)表明,MgH2+10%(質(zhì)量分?jǐn)?shù))納米Ni 復(fù)合材料在250 ℃下可在10 min 內(nèi)脫附6.1%H2。隨著催化劑用量的增加,MgH2+納米Ni復(fù)合材料的脫氫速率明顯提高,但飽和儲(chǔ)氫量會(huì)有一定衰減。結(jié)果表明,通過改變催化劑用量,可以進(jìn)一步調(diào)控Ni 的催化作用。
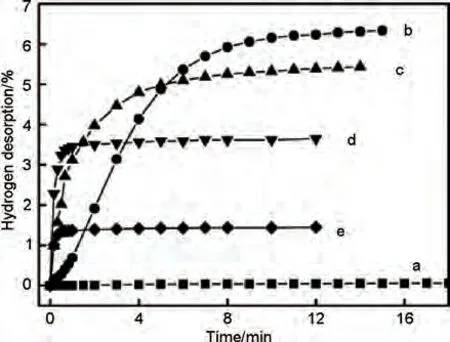
圖22 (a) 球磨MgH2;(b) MgH2+10%Ni;(c) MgH2+25%Ni;(d) MgH2+50%Ni和(e) MgH2+80%Ni在523 K時(shí)的等溫脫氫曲線[116]Fig.22 Hydrogen desorption curves of the (a) MgH2 milled for 2 h; (b) MgH2+10%Ni; (c) MgH2+25% Ni;(d) MgH2+50% Ni, and (e) MgH2+80% Ni at 523 K under an initial pressure of 0.01bar of H2[116]
Yang 等[117]研究了Ni 顆粒尺寸對MgH2氫解吸性能的影響。結(jié)果表明,僅有2%(原子分?jǐn)?shù))細(xì)鎳顆粒引入的MgH2在200 ℃時(shí)即可快速脫氫,當(dāng)溫度升至340 ℃時(shí),脫氫量高達(dá)6.5%。然而,DSC曲線(見圖23)表明,MgH2-2%(原子分?jǐn)?shù))Ni90(90 為Ni顆粒粒徑)復(fù)合材料的峰值脫氫溫度約為280 ℃,僅比MgH2-2%(原子分?jǐn)?shù))Ni200和MgH2-2%(原子分?jǐn)?shù))Ni100復(fù)合材料的峰值溫度低約10 ℃。具有8%(原子分?jǐn)?shù))微尺寸Ni 的MgH2粉末在310 ℃和390 ℃下分兩步釋放氫氣:第二步的脫氫溫度(390 ℃)非常接近研磨態(tài)MgH2。因此,第二個(gè)吸熱峰的存在可能是由于Ni 的不均勻分布使一些MgH2顆粒沒有被完全催化導(dǎo)致的。即催化劑尺寸的減小并不是改善儲(chǔ)氫材料性能的關(guān)鍵,催化劑在MgH2顆粒上的位點(diǎn)分布和密度才是提高M(jìn)gH2吸脫氫性能的關(guān)鍵因素。
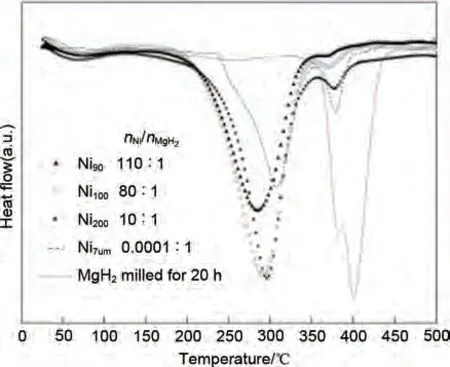
圖23 簡單球磨20 h的MgH2和不同類型的Ni(90 nm、100 nm、200 nm、7 μm)的DSC曲線[117]Fig.23 Comparison of the effects of different Ni particle sizes on the desorption properties of pre-milled MgH2 by simply mixed 20 h-pre-milled MgH2 and different types Ni (90 nm, 100 nm, 200 nm and 7 μm)[117]
針對過渡金屬Fe,F(xiàn)e是地球上最重要的元素,在自然界中廣泛分布。近年來,其優(yōu)異的催化性能受到了學(xué)者們的廣泛關(guān)注。Bassetti 等[118]采用高能球磨法制備了MgH2-Fe 納米復(fù)合材料。對不同F(xiàn)e含量的MgH2進(jìn)行球磨后發(fā)現(xiàn),微晶尺寸、粉末粒徑、催化劑濃度和分散性是影響脫氫動(dòng)力學(xué)的關(guān)鍵因素。DSC 曲線分析表明,MgH2-Fe 的脫氫溫度基本在220 ℃左右。添加10%(質(zhì)量分?jǐn)?shù))Fe 時(shí)在300 ℃下、600 s 內(nèi)釋放約5%(質(zhì)量分?jǐn)?shù))H2,具有最佳的催化效果。
眾所周知,F(xiàn)e原子可以通過吸附和取代Mg來提高儲(chǔ)氫性能,但其潛在機(jī)理尚不清楚。為了解決此問題,Chen 等[119]基于密度泛函理論計(jì)算研究了Fe 原子不同引入形式對Mg 晶體性質(zhì)的影響(圖24)。通過模擬Fe 原子的吸附、解離和擴(kuò)散過程,發(fā)現(xiàn)Fe原子能增強(qiáng)氫分子在Mg(0001)表面的吸附性能。氫解離計(jì)算表明,F(xiàn)e 對H2解離的催化作用可以歸結(jié)為被吸附Fe 原子的橋接作用,使電子從H2成鍵軌道轉(zhuǎn)移到反成鍵軌道,從而削弱了H—H鍵。為了更好地了解Fe對Mg基體系氫吸附動(dòng)力學(xué)的影響,Antiqueira等[120]采用10 h和24 h高能球磨技術(shù)對Mg-8%(質(zhì)量分?jǐn)?shù))Fe 納米復(fù)合材料進(jìn)行了研磨。這兩種納米復(fù)合材料都具有非常快的吸收/解吸動(dòng)力學(xué)。在350 ℃時(shí),2 min內(nèi)即可完全脫氫,10 min 內(nèi)實(shí)現(xiàn)飽和吸氫。實(shí)驗(yàn)結(jié)果進(jìn)一步表明,當(dāng)磨粉時(shí)間延長至24 h 時(shí),出現(xiàn)了Mg2FeH6與MgH2含量較高的混合物,Mg2FeH6的存在降低了復(fù)合材料的儲(chǔ)氫能力,使解吸溫度略有上升。因此,控制球磨時(shí)間是改善儲(chǔ)氫材料性能的關(guān)鍵。
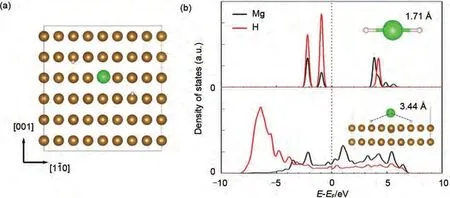
圖24 MgH2在Fe(110)晶面上的吸附幾何形狀(棕色、綠色和白色球分別代表鐵、鎂和氫)(a);Mg和H在Fe(110)晶面上吸附之前(上)和吸附后(下)的預(yù)測態(tài)密度(PDOS)(b)[108]Fig.24 The adsorption geometry of MgH2 on Fe (110)surface in top view (a); The brown, green, and white ball represent Fe, Mg, and H, respectively, The projected density of states (PDOS)of Mg and H of MgH2 before (upper)and after (lower)adsorption on Fe (110)surface (b) [108]
Zhang等[121]采用濕法化學(xué)球磨法制備了厚度約為30 nm 的二維層狀Fe 納米片,并研究了其對MgH2的影響。MgH2+5%(質(zhì)量分?jǐn)?shù))Fe 的納米復(fù)合材料具有最佳的吸氫和解吸性能。脫氫溫度為182.1 ℃,吸氫溫度為75 ℃。此外,在200 ℃下,10 分鐘內(nèi)可吸收6%(質(zhì)量分?jǐn)?shù))H2。經(jīng)過幾次循環(huán)測試,氫含量保持在5%(質(zhì)量分?jǐn)?shù))左右。進(jìn)一步分析表明,F(xiàn)e 納米片的加入可以削弱Mg—H 鍵從而提高M(jìn)gH2的脫氫性能(圖24)。
綜上所述,將具有優(yōu)異催化效果的過渡金屬單質(zhì)引入MgH2中可以有效改善吸脫氫性能,削弱Mg—H 鍵,降低儲(chǔ)氫材料的活化能。但是單一過渡金屬的引入,無法實(shí)現(xiàn)多元素的協(xié)同催化效果,另外過渡金屬單質(zhì)硬度較高,在一定程度上降低了球磨效率,限制了它的應(yīng)用。
2.3.2 過渡金屬氧化物改性
與金屬單質(zhì)相比,金屬氧化物具有更高的脆性,通過球磨技術(shù)使其更容易分散在MgH2基體中,引入過渡金屬氧化物同樣可以有效地催化MgH2的吸放氫反應(yīng)[122-123]。過渡金屬氧化物根據(jù)所含元素可分為二元、三元兩大類。
對于二元過渡金屬氧化物,早在1991 年Terzieva 等[124]就報(bào)道了Fe2O3和MnO2能夠提升儲(chǔ)氫材料的吸放氫動(dòng)力學(xué)性能。研究發(fā)現(xiàn),金屬氧化物部分還原為金屬顆粒,這些金屬可加快氫分子的分解,從而促進(jìn)體系的動(dòng)力學(xué)性能。隨后,2001 年Oelerich 等[125]通過簡單球磨的方法將各種一元過渡金屬氧化物(Sc2O3、TiO2、V2O5、Cr2O3、Mn2O3、Fe3O4、CuO/Al2O3和SiO2)引 入MgH2中,并探討了它們對Mg基儲(chǔ)氫材料吸脫氫性能的影響。在這些氧化物中,添加CuO的MgH2復(fù)合材料可吸氫5.5%(質(zhì)量分?jǐn)?shù))H2,在300 ℃真空條件下10 min之內(nèi)實(shí)現(xiàn)完全脫氫,添加V2O5和Fe3O4的氫化鎂在5 min 之內(nèi)就可完成放氫。高價(jià)態(tài)Nb2O5和ZrO2對MgH2體系的吸放氫性能也有顯著提升。
然而,由于簡單球磨法制備的TiO2納米片在循環(huán)過程中容易發(fā)生團(tuán)聚,嚴(yán)重減少催化活性位點(diǎn)[126]。為了解決這一問題,鄒建新團(tuán)隊(duì)[127]首次嘗試?yán)眉{米尺寸效應(yīng)和催化劑添加效應(yīng),通過溶劑熱法設(shè)計(jì)了一種新型的自組裝MgH2/TiO2異質(zhì)結(jié)構(gòu)納米復(fù)合材料。該復(fù)合材料的催化機(jī)理如圖25 所示:①高比表面積的TiO2納米片為氫的擴(kuò)散提供了更多的通道,并為MgH2/Mg提供了大量成核位點(diǎn)。②納米MgH2均勻負(fù)載在TiO2納米片上,顯著抑制了納米MgH2在吸氫/脫氫過程中的生長和團(tuán)聚,確保了良好的循環(huán)穩(wěn)定性。③大量氧空位顯著提高了TiO2的導(dǎo)電性,并為電子和氫的運(yùn)輸提供了額外的活性位點(diǎn),進(jìn)一步改善吸脫氫性能。④多價(jià)Ti基催化劑組成的多相界面為MgH2/Mg 提供更多的氫擴(kuò)散途徑和成核位點(diǎn)。性能方面:MgH2/TiO2的起始脫氫溫度降至180 ℃。在300 ℃時(shí),MgH2/TiO2的脫氫速率為2.116%(質(zhì)量分?jǐn)?shù))/min,是相同條件下純MgH2的35倍。
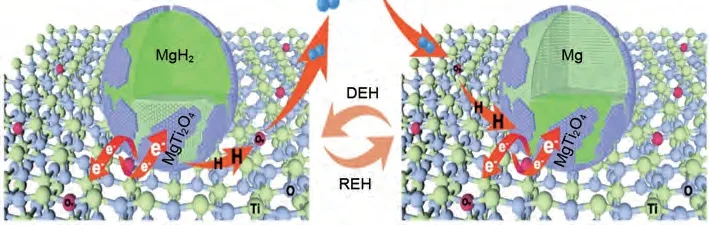
圖25 MgH2/TiO2異質(zhì)結(jié)構(gòu)吸氫和脫氫機(jī)理示意圖[127]Fig.25 Schematic diagram of the hydrogenation and dehydrogenation mechanisms of MgH2/TiO2 heterostructure[127]
針對三元過渡金屬氧化物,第一種是以MgH2-TM(O)/TMO(TM為過渡金屬)形式復(fù)合。
朱云峰團(tuán)隊(duì)等[128]首次通過層狀雙金屬氫氧化物(LDH)前驅(qū)體在氫氣和氬氣氛圍下煅燒分別制備了層狀Ni/Al2O3和NiO/Al2O3。對比發(fā)現(xiàn)Ni/Al2O3對MgH2的吸氫性能具有更加優(yōu)異的催化作用。如圖26 所示,TPD 曲線[圖26(a)]表明,MgH2-Ni/Al2O3的起始脫氫溫度(190 ℃)遠(yuǎn)低于MgH2-NiO/Al2O3(240 ℃)和球磨后的MgH2(298 ℃),說明Ni/Al2O3對脫氫的催化活性優(yōu)于NiO/Al2O3。等溫脫氫動(dòng)力學(xué)中也存在類似現(xiàn)象。在275 ℃時(shí),MgH2-5%(質(zhì)量分?jǐn)?shù))Ni/Al2O3脫氫動(dòng)力學(xué)更快,在3500 s內(nèi)脫附5.6%(質(zhì)量分?jǐn)?shù))H2,MgH2-5%(質(zhì)量分?jǐn)?shù))Ni/Al2O3脫附4.6%(質(zhì)量分?jǐn)?shù))H2,球磨后的MgH2幾乎無法脫氫[圖26(b)]。在250 ℃和300 ℃下[圖26(c)、(d)],MgH2-Ni/Al2O 比MgH2-NiO/Al2O3展現(xiàn)出更好的脫氫動(dòng)力學(xué)和更高的儲(chǔ)氫量。通過擬合不同的脫氫動(dòng)力學(xué)模型發(fā)現(xiàn)[圖26(e)],斜率最接近于1的動(dòng)力學(xué)模型為F1模型(一級動(dòng)力學(xué)),即脫氫速控步為氫的重組[129]。通過Arrhenius方程計(jì)算的MgH2-5%(質(zhì)量分?jǐn)?shù))Ni/Al2O3的脫氫活化能Ea(72.6 kJ/mol)[圖26(f)]遠(yuǎn)低于MgH2(160 kJ/mol)。最后,結(jié)合實(shí)驗(yàn)和DFT計(jì)算證明,該復(fù)合材料氫解吸動(dòng)力學(xué)增強(qiáng)主要是由于Ni/Al2O3作為電子受體捕獲Mg—H鍵上的電子,造成MgH2的失穩(wěn)效應(yīng),導(dǎo)致Mg—H鍵強(qiáng)度減弱,從而改善儲(chǔ)氫性能。
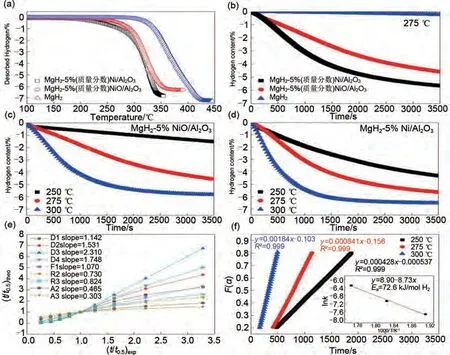
圖26 (a) MgH2、MgH2-5%(質(zhì)量分?jǐn)?shù))Ni/Al2O3和MgH2-5%(質(zhì)量分?jǐn)?shù))NiO/Al2O3的程序升溫脫附曲線以及(b) 275 ℃下的等溫脫氫曲線;(c) MgH2-5%(質(zhì)量分?jǐn)?shù))NiO/Al2O3和(d) MgH2-5%(質(zhì)量分?jǐn)?shù))Ni/Al2O3在250 ℃、275 ℃和300 ℃下的等溫脫氫曲線;(e) MgH2-5%(質(zhì)量分?jǐn)?shù))Ni/Al2O3對應(yīng)的固相反應(yīng)機(jī)理模型和速率控制步驟;(f) 隨時(shí)間變化的動(dòng)力學(xué)模型以及由Arrhenius方程得到的隨溫度變化的速率常數(shù)圖[128]Fig.26 (a)Temperature-programmed-desorption and (b) isothermal dehydrogenation curves at 275 ℃ of MgH2,MgH2-5%Ni/Al2O3 and MgH2-5%NiO/Al2O3 ; isothermal dehydrogenation curves of (c)MgH2-5%NiO/Al2O3 and (d)MgH2-5%Ni/Al2O3 at different temperatures of 250 ℃, 275 ℃ and 300 ℃; (e)the corresponding solid-state reaction mechanism model and rate-controlling step of MgH2-5%Ni/Al2O3; (f)time dependence of the kinetic modeling equation F(α)and the plot for the temperature-dependent rate constant k, obtained by the Arrhenius equation[128]
武英團(tuán)隊(duì)[130]將TiO2引入Sc2O3中,采用溶膠-凝膠法和煅燒法合成了Sc2O3/TiO2催化劑,并將其加入MgH2中以改善MgH2的脫氫動(dòng)力學(xué)性能。計(jì)算得到MgH2-5%(質(zhì)量分?jǐn)?shù))Sc2O3/TiO2復(fù)合材料的脫氫活化能為77.8 kJ/mol,比商用MgH2(160 kJ/mol)低一半。此外,當(dāng)在MgH2中加入5%(質(zhì)量分?jǐn)?shù))的Sc2O3/TiO2催化劑后,復(fù)合材料儲(chǔ)氫性能顯著提高,在300 ℃下1000 s內(nèi)釋放6.21%(質(zhì)量分?jǐn)?shù))H2,并且可以在200 ℃下3000 s 內(nèi)吸收6.08%(質(zhì)量分?jǐn)?shù))H2,性能遠(yuǎn)超Mg/MgH2。優(yōu)異的儲(chǔ)氫性能可歸因于均勻分布在Mg顆粒表面的Sc和Ti的協(xié)同催化效應(yīng)和對Mg/MgH2團(tuán)聚粗化的抑制效應(yīng)(見圖27)。
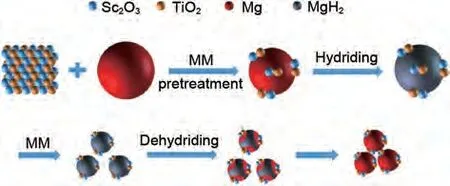
圖27 MgH2@5%(質(zhì)量分?jǐn)?shù))Sc2O3/TiO2復(fù)合材料在吸氫--脫氫過程中的微觀結(jié)構(gòu)演變示意圖[130]Fig.27 Schematic diagram of the microstructural evolution for the MgH2@5%Sc2O3/TiO2 composite during the hydrogenation-dehydrogenation process [130]
第二種是以MgH2-TMTMO形式復(fù)合。
Sazelee等[131]通過高溫固相法合成了BaFe12O19,結(jié)果表明,MgH2+10%(質(zhì)量分?jǐn)?shù))BaFe12O19樣品在約270 ℃開始脫氫,比研磨后的MgH2低約70 ℃。MgH2+10%(質(zhì)量分?jǐn)?shù))BaFe12O19復(fù)合材料能夠在150 ℃下10 分鐘內(nèi)吸附4.3%(質(zhì)量分?jǐn)?shù))H2,而研磨后的MgH2僅能吸附3.5%(質(zhì)量分?jǐn)?shù))H2。此外,摻雜BaFe12O19的MgH2材料的脫氫活化能為115 kJ/mol, 低 于 研 磨 的MgH2(141 kJ/mol)。Zhang 等[132]結(jié)合共沉淀法和水熱法合成了LiCoO2納米片,發(fā)現(xiàn)它對MgH2的儲(chǔ)氫性能具有顯著的催化作用。MgH2-LiCoO2體系的初始脫氫溫度降至180 ℃,250 ℃下60 min內(nèi)脫氫量高達(dá)5.5%(質(zhì)量分?jǐn)?shù))。脫氫活化能降低至(48.5±0.4) kJ/mol,相比MgH2(81.4±5.6 kJ/mol)降低了40.4%。分析表明,LiCoO2在MgH2中均勻分布,并在循環(huán)過程中自組裝成大量Mg2Co-Mg2CoH5“納米氫泵”,從而加速了氫的擴(kuò)散速率,提高了儲(chǔ)氫性能(見圖28)。
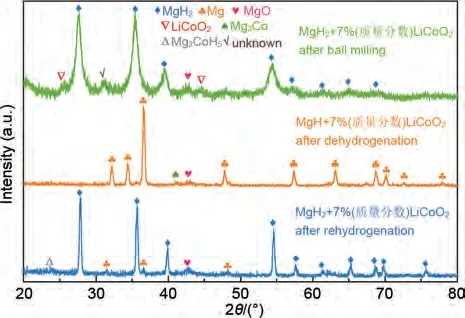
圖28 MgH2+7% LiCoO2復(fù)合材料球磨后、脫氫后和再吸氫后的XRD譜圖[131]Fig.28 XRD patterns of MgH2+7% LiCoO2 composite:after ball milling, after dehydrogenation and after rehydrogenation [131]
鑒于Mg2Co 和Mg2CoH5之間的可逆相變具有的“氫泵”效應(yīng)可有效改善鎂基儲(chǔ)氫材料的吸脫氫性能。潘復(fù)生團(tuán)隊(duì)[133]通過水熱和煅燒兩步方法成功合成了厚度為60~100 nm 的多層頁片狀MnCo2O4.5納米顆粒,以探索二元鈷和錳基氧化物對MgH2的催化作用。MgH2-6%(質(zhì)量分?jǐn)?shù))MnCo2O4.5復(fù)合材料的初始脫氫溫度降至285 ℃,在325 ℃下4 min內(nèi)完全釋放6.4%(質(zhì)量分?jǐn)?shù))H2。脫氫后的MgH2-6%(質(zhì)量分?jǐn)?shù))MnCo2O4.5復(fù)合材料在150 ℃下30 min 內(nèi)吸收4.43%(質(zhì)量分?jǐn)?shù))H2,吸脫氫性能相比球磨態(tài)MgH2有了顯著改善[見圖29(a)~(d)],復(fù)合材料經(jīng)30 次吸脫氫循環(huán)后吸氫量僅衰減了0.75%(質(zhì)量分?jǐn)?shù)),脫氫量基本維持在6.24%(質(zhì)量分?jǐn)?shù))左右,展現(xiàn)出了優(yōu)異的循環(huán)儲(chǔ)氫性能[見圖29(e)、(f)]。一些常見的MgH2-三元過渡金屬氧化物復(fù)合材料的儲(chǔ)氫性能如表5所示。
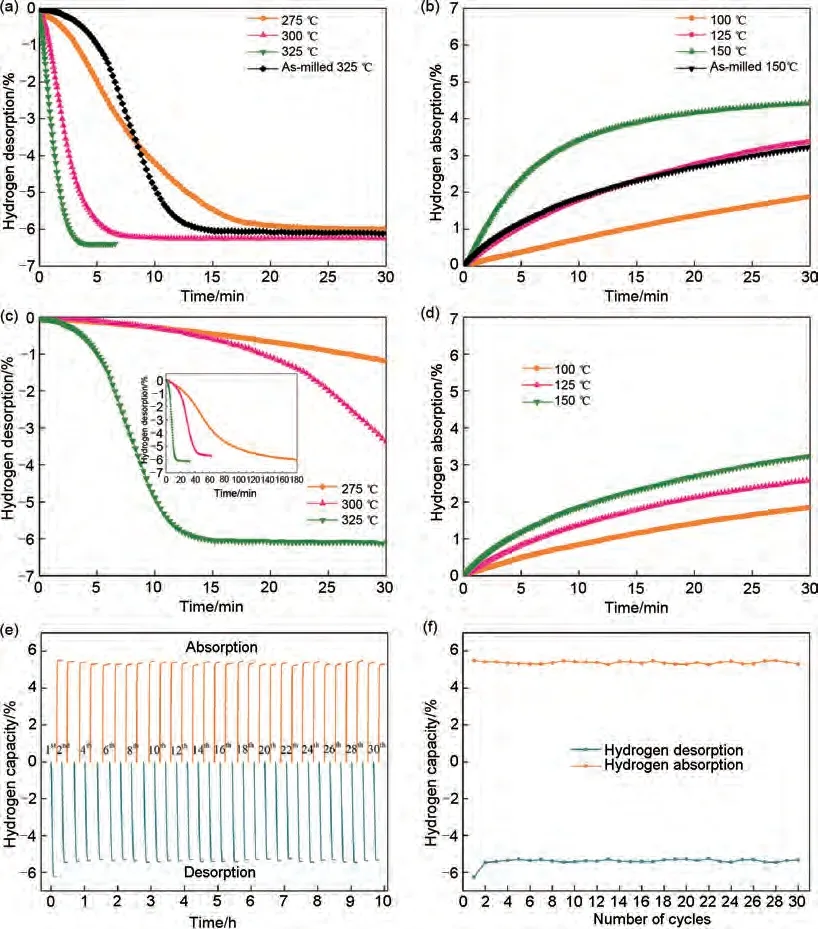
圖29 (a) MgH2-6%(質(zhì)量分?jǐn)?shù))MnCo2O4.5和(c)碾磨態(tài)MgH2的等溫脫氫曲線;(b)MgH2-6%(質(zhì)量分?jǐn)?shù))MnCo2O4.5和(d)碾磨態(tài)MgH2的等溫吸氫曲線;(e)MgH2-6%(質(zhì)量分?jǐn)?shù))MnCo2O4.5在325 ℃時(shí)的脫氫循環(huán)曲線;(f)儲(chǔ)氫容量的衰變曲線[120]Fig.29 (a)Isothermal dehydrogenation curves of MgH2-6 wt% MnCo2O4.5 and (c)as-milled MgH2; (b)isothermal hydrogenation curves of MgH2-6 wt% MnCo2O4.5 and (d)as-milled MgH2; (e)de/hydrogenation cycle curves of MgH2-6 wt% MnCo2O4.5 at 325 ℃, and (f)decay curves of hydrogen storage capacity[120]
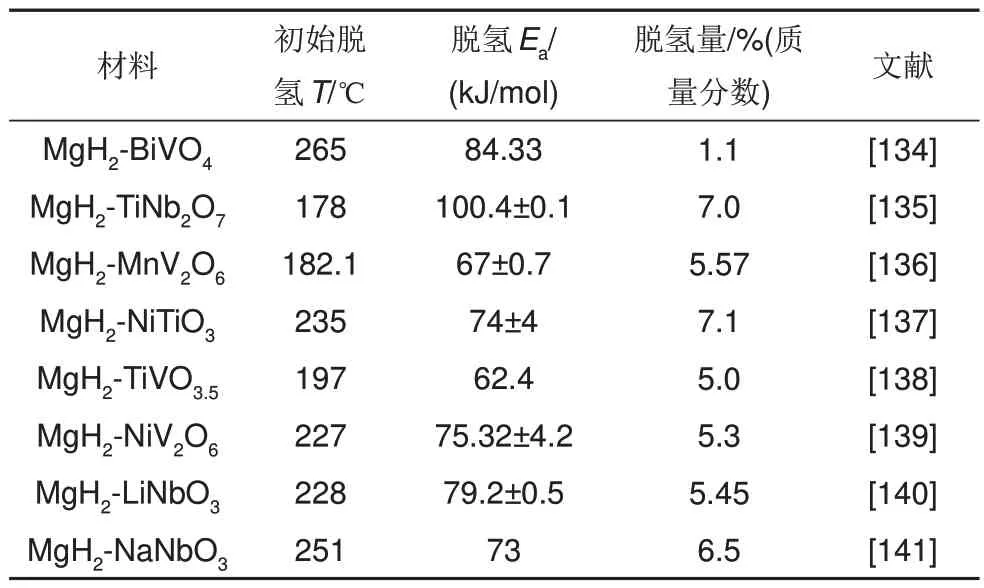
表5 一些MgH2-三元過渡金屬氧化物的儲(chǔ)氫性能Table 5 Hydrogen storage properties of some MgH2-ternary transition metal oxides
綜上所述,多元過渡金屬氧化物可以展現(xiàn)出比單一過渡金屬更加優(yōu)異的催化效果,氧空位的引入可以大大加快電子轉(zhuǎn)移,進(jìn)一步改善儲(chǔ)氫材料的熱力學(xué)/動(dòng)力學(xué)性能。
2.3.3 其他金屬化合物
除金屬氧化物外,氟化物、硫化物和金屬-有機(jī)骨架化合物(MOFs)等其他金屬化合物由于其特殊的物理和化學(xué)性質(zhì)而表現(xiàn)出顯著的催化活性,其對Mg/MgH2體系的催化作用也受到廣泛關(guān)注。
針對氟化物,Lin 等[142]研究了球磨法制備的不同價(jià)態(tài)氟化鈰摻雜MgH2的氫解吸性能。研究發(fā)現(xiàn),當(dāng)摻雜2%(摩爾分?jǐn)?shù))CeF4后,MgH2的氫解吸溫度和表觀活化能顯著降低,這可能是由于CeF4/MgH2界面上形成的新相Ce-F-Mg 和高價(jià)Ce 陽離子促進(jìn)了電子轉(zhuǎn)移所導(dǎo)致的。此外,MgH2-CeF4的解吸活化能從160 kJ/mol降至110 kJ/mol。Mao等[143]采用電弧等離子體法制備核殼結(jié)構(gòu)Mg-MFx(M=V、Ni、La、Ce)納米復(fù)合材料。其中,Mg-NiF2復(fù)合材料在相對較低的溫度下表現(xiàn)出最佳的吸氫性能,在100 ℃下2 h 內(nèi)吸氫量達(dá)到3.26%(質(zhì)量分?jǐn)?shù)),在473 K 下60 s 內(nèi)吸氫量達(dá)到3.85%(質(zhì)量分?jǐn)?shù)),優(yōu)異的吸氫性能主要是由于電弧蒸發(fā)和冷凝過程中Mg粒子上形成了Mg2Ni和MgF2。Mg-VF3復(fù)合材料具有最低的初始脫氫溫度(674.2 K),MgF2和金屬氧化物覆蓋在Mg 顆粒上的核殼結(jié)構(gòu)和較少的團(tuán)聚傾向是儲(chǔ)氫性能改善的主要原因。
與單陽離子氟化物相比,由于過渡金屬與鉀元素的協(xié)同催化作用,雙陽離子氟化物也被廣泛用作鎂基儲(chǔ)氫材料的優(yōu)良催化劑。Sulaiman 等[144]將K2NiF6添加到MgH2中以改善其脫氫特性。結(jié)果發(fā)現(xiàn),MgH2-5%(質(zhì)量分?jǐn)?shù))K2NiF6材料在260 ℃下可以脫氫,KH、KF 和Mg2Ni 催化相的出現(xiàn)共同改善了MgH2的儲(chǔ)氫行為(圖30)。Yan等[145]首次研究了新型雙陽離子金屬氟化物K2TaF7對MgH2儲(chǔ)氫特性的催化影響。在只有1%(質(zhì)量分?jǐn)?shù))K2TaF7摻雜劑的情況下,MgH2的初始脫氫溫度降低了約130 ℃,總脫氫量超過7.3%(質(zhì)量分?jǐn)?shù))。MgH2-1%(質(zhì)量分?jǐn)?shù))K2TaF7復(fù)合材料的解吸活化能降低至(107.2±1.2) kJ/mol。在190 ℃,MgH2-1%(質(zhì)量分?jǐn)?shù))K2TaF7樣品吸氫量達(dá)到6.56%(質(zhì)量分?jǐn)?shù)),而MgH2吸氫量只有3.45%(質(zhì)量分?jǐn)?shù))。研究表明,K2TaF7在脫氫過程中可以與MgH2反應(yīng),產(chǎn)生共生氫化物KMgH3和TaH0.8,在氫的釋放和吸收過程中發(fā)揮氫泵的作用(圖31)。
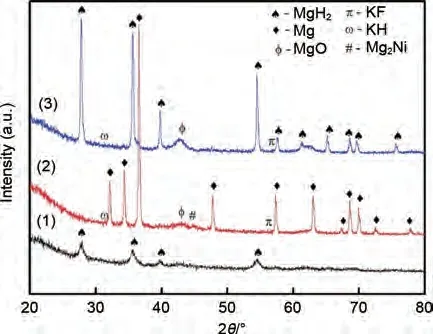
圖30 MgH2+5%(質(zhì)量分?jǐn)?shù))K2NiF6球磨1 h、450 ℃脫氫和320 ℃吸氫后的XRD譜圖[144]Fig.30 XRD patterns of MgH2+5%K2NiF6 composite:after ball milling, after dehydrogenation and after rehydrogenation [144]
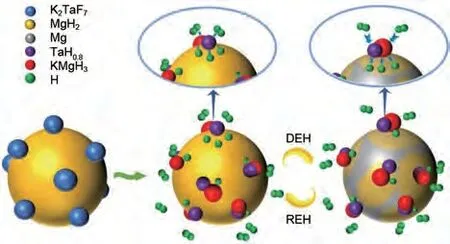
圖31 K2TaF7對MgH2的催化機(jī)理圖[145]Fig.31 Schematic illustration of the catalytic mechanism of the K2TaF7-catalyzed MgH2 composite[145]
針對硫化物,Wang等[146]通過球磨將過渡金屬硫化物(TiS2、NbS2、MoS2、MnS、CoS2、CuS)作為催化劑來改善MgH2的儲(chǔ)氫性能。結(jié)果表明,這些硫化物均能顯著提高M(jìn)gH2的氫解吸動(dòng)力學(xué)。MgH2-TiS2的吸氫和脫氫動(dòng)力學(xué)最好,MgH2-TiS2的初始脫氫溫度約為204 ℃,比MgH2低約126 ℃。脫氫活化能降低至50.8 kJ/mol。硫化物的有益催化作用可歸因于原位形成的MgS、TiH2、NbH、Mo、Mn、Mg2CoH5和MgCu2相。
基于新型雙陽離子金屬硫化物,韓樹民團(tuán)隊(duì)[147]設(shè)計(jì)了一種中空球囊結(jié)構(gòu)的三元過渡金屬硫化物FeNi2S4作為MgH2的催化劑。值得注意的是,MgH2-FeNi2S4復(fù)合材料中活性物質(zhì)Mg2Ni/Mg2NiH4、MgS 和Fe 的協(xié)同催化作用顯著提高了MgH2的脫氫/加氫性能(圖32)。MgH2-FeNi2S4復(fù)合材料在373 K下1 h內(nèi)的吸氫量達(dá)到4.02%(質(zhì)量分?jǐn)?shù)),與球磨態(tài)MgH2(0.67%,質(zhì)量分?jǐn)?shù))形成鮮明對比。脫氫過程中,MgH2-FeNi2S4復(fù)合材料的初始脫氫溫度比球磨態(tài)MgH2低80 K,脫氫活化能比MgH2(161.2 kJ/mol)降低了95.7 kJ/mol。
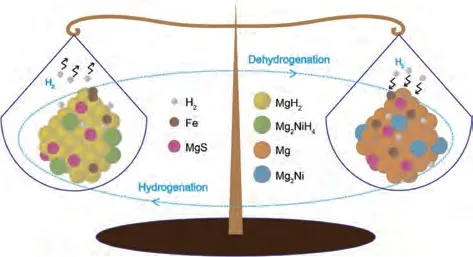
圖32 MgH2-FeNi2S4復(fù)合材料的加氫/脫氫過程示意圖 [147]Fig.32 Schematic diagram of the hydrogenation/dehydrogenation processes of the MgH2-FeNi2S4 composite [147]
除了氟化物與硫化物,金屬-有機(jī)骨架化合物在改善鎂基儲(chǔ)氫材料性能方面也得到了深入的研究。金屬-有機(jī)骨架(MOFs)化合物是以金屬離子為連接中心,有機(jī)配體為橋聯(lián)體,通過配位鍵、氫鍵、π-π鍵、范德華力等作用力,自組裝而成的具有周期性的多孔配位聚合物[148]。由于具有充足的金屬中心、較大的比表面積、豐富的孔道結(jié)構(gòu)、可設(shè)計(jì)的拓?fù)鋯卧约办`活可調(diào)的組成成分,MOFs的應(yīng)用研究延伸至眾多領(lǐng)域[149-151]。2003 年,美國密歇根大學(xué)的Yaghi 教授等[152]首次報(bào)道了MOF-5 的儲(chǔ)氫性能,MOF-5 是由Zn2+鹽和1, 4-對苯二甲酸有機(jī)配體合成的多孔八面體結(jié)構(gòu)骨架,經(jīng)過測試發(fā)現(xiàn)在77 K、1 bar(1 bar=100 kPa)外界條件下,MOF-5 的儲(chǔ)氫量為1.3%(質(zhì)量分?jǐn)?shù))。但正如引言中所提到的,MOF 材料只能在極低溫度下實(shí)現(xiàn)儲(chǔ)氫,這與國際能源機(jī)構(gòu)規(guī)定的在室溫下實(shí)現(xiàn)高效儲(chǔ)氫的要求相去甚遠(yuǎn),但鑒于其比表面積大、孔隙率高、活性金屬位點(diǎn)多等優(yōu)點(diǎn),近年來作為催化劑、催化劑載體被廣泛應(yīng)用于改善鎂基儲(chǔ)氫材料儲(chǔ)氫性能的研究當(dāng)中[153]。
MOF材料的中心原子大多為過渡金屬(Co、Ni等)或具有催化效果的非過渡金屬(Mg、Zn 等),因此其自身即可作為催化劑用于改善Mg的儲(chǔ)氫性能。
MOF 材料ZIF-8、ZIF-67 和MOF-74 的中心原子分別為Zn、Co和Mg,Wang等[154]采用沉積還原法制備了Mg/MOF(MOF=ZIF-8、ZIF-67、MOF-74)儲(chǔ)氫復(fù)合材料,如圖33(a)所示,在175 ℃下Mg/ZIF-67、Mg/ZIF-8 和Mg/MOF-74 分別在2051 s、3759 s和3966 s內(nèi)吸收2%(質(zhì)量分?jǐn)?shù))H2,分別比未添加MOF 材料的Mg 納米顆粒快3124 s、1708 s 和1915 s,吸氫速度的提升很好地證明了Mg-MOF復(fù)合體系的優(yōu)異催化效果。在300 ℃時(shí)研究了材料的脫氫動(dòng)力學(xué)性能[見圖33(b)],5000 s內(nèi)Mg、Mg/MOF-74、Mg/ZIF-8 和Mg/ZIF-67 分別釋放了0.6%(質(zhì)量分?jǐn)?shù))、1.2%(質(zhì)量分?jǐn)?shù))、2.7%(質(zhì)量分?jǐn)?shù))和3.7%(質(zhì)量分?jǐn)?shù))H2,Mg/ZIF-67具有最為可觀的脫氫量。此外,Mg/ZIF-67復(fù)合材料在350 ℃時(shí)最大儲(chǔ)氫量達(dá)到了5.30%(質(zhì)量分?jǐn)?shù)),即使在100次氫氣吸收/解吸循環(huán)后,吸脫氫量沒有任何衰減。
圖33 Mg、Mg/ZIF-8、Mg/ZIF-67和Mg/MOF-74復(fù)合材料的等溫氫化曲線[154]:(a)175 ℃吸氫過程;(b)300 ℃脫氫過程Fig.33 Isothermal hydriding curves of the Mg, Mg/ZIF-8, Mg/ZIF-67, and Mg/MOF-74composites [154]: (a)hydrogenation process at 175 ℃; (b)dehydrogenation process at 300 ℃
以過渡金屬作為中心原子制備出的MOF 可以大大增加比表面積,產(chǎn)生更多的吸脫氫擴(kuò)散通道,從而大大改善儲(chǔ)氫性能。朱云峰團(tuán)隊(duì)等[155]通過簡單的水熱反應(yīng)首次制備了結(jié)構(gòu)穩(wěn)定的新型花狀Ni-MOF材料,并將其引入MgH2中,研究表明該材料在提高M(jìn)gH2的儲(chǔ)氫性能方面表現(xiàn)出優(yōu)異的催化活性。MgH2-5%(質(zhì)量分?jǐn)?shù))Ni-MOF 的峰值脫氫溫度比純MgH2低78 ℃。該復(fù)合材料在300 ℃下600 s內(nèi)釋放6.4%(質(zhì)量分?jǐn)?shù))H2,并在150 ℃吸附約5.7%(質(zhì)量分?jǐn)?shù))H2。花狀Ni-MOF 的高催化活性可歸因于原位生成的Mg2Ni/Mg2NiH4、MgO 納米顆粒、無定形C和剩余的層狀Ni-MOF的結(jié)合作用(見圖34)。
圖34 (a) 明場TEM圖; (b)SAED圖; (c)、(d) HRTEM圖像; (e)~(i) 暗場圖和11次氫化后MgH2-5%(質(zhì)量分?jǐn)?shù))Ni-MOF的EDS圖[155]Fig.34 (a)Typical bright field TEM micrograph; (b)the corresponding SAED pattern; (c)、(d)the HRTEM images, (e)—(i)the dark field micrographs and the corresponding elemental mappings of MgH2-5% Ni-MOF after 11th hydrogenation [155]
此外,具有較大比表面積和豐富多孔結(jié)構(gòu)的MOF 材料具有大量的新鮮表面,可作為過渡金屬和Mg的高效載體,通過過渡金屬與MOF材料的協(xié)同作用改善鎂基材料的儲(chǔ)氫性能。
郭進(jìn)團(tuán)隊(duì)[156]以ZIF-67 為催化劑載體,采用Mg和過渡金屬Nb 共沉積還原的方法成功制備了MgNb/ZIF-67 復(fù)合材料,并研究了Nb 和ZIF-67 對Mg 儲(chǔ)氫性能的協(xié)同作用。如圖35(a)所示,在175 ℃下,MgNb 和MgNb/ZIF-67 吸附2%(質(zhì)量分?jǐn)?shù))H2分別需要23 min 和10 min,MgNb/ZIF-67 具有更快的吸氫速率,在275 ℃的脫氫過程中[圖35(b)],MgNb/ZIF-67僅需要8 min就可釋放出3%(質(zhì)量分?jǐn)?shù))H2,而MgNb 需要23 min 才能達(dá)到相同的脫氫量。MgNb/ZIF-67更為迅速的吸脫氫速率證實(shí)了過渡金屬與MOF 材料之間存在協(xié)同催化作用。但是制備出的Mg顆粒存在嚴(yán)重的團(tuán)聚現(xiàn)象[見圖36(a)],而MgNb/ZIF-67 具有豐富的多孔結(jié)構(gòu)且無明顯團(tuán)聚現(xiàn)象[見圖36(b)],由100次循環(huán)后復(fù)合材料的TEM圖[圖36(c)]可看出,ZIF-67中心原子Co所延伸的晶面負(fù)載了大量的晶格Mg,剩余的少量Mg、Nb 和部分氧化的NbO 緊密接觸,形成了CoMg2-Mg(Nb)核殼結(jié)構(gòu),該結(jié)構(gòu)具有納米限域的效果,保證了MgNb/ZIF-67穩(wěn)定的循環(huán)儲(chǔ)氫性能。
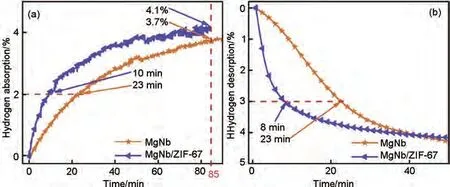
圖35 MgNb和MgNb/ZIF-67納米復(fù)合材料在(a)175 ℃和(b)275 ℃下的等溫吸/脫氫曲線[156]Fig.35 Isothermal hydrogenation/dehydrogenation curves of MgNb and MgNb/ZIF-67 nanocomposites at (a)175 ℃ and (b)275 ℃ [156]

圖36 (a)、(b) Mg和MgNb/ZIF-67納米復(fù)合材料的SEM圖; (c)~(e)MgNb/ZIF-67 100次吸/脫氫循環(huán)后的TEM圖像和SAED圖[156]Fig.36 (a)、(b) SEM micrographs of the Mg and MgNb/ZIF-67 nanocomposites (c)—(e)TEM images and SAED of MgNb/ZIF-67 after 100 hydrogen absorption/desorption cycles [156]
Zhang 等[157]通過水熱反應(yīng)和隨后的煅燒過程,成功將Nb2O5納米粒子負(fù)載在了菱形十二面體金屬有機(jī)骨架上,該納米粒子的平均粒徑為10 nm(圖37)。如圖37 所示,復(fù)合了7%(質(zhì)量分?jǐn)?shù))Nb2O5@MOF 的MgH2在181.9 ℃開始脫氫,在275 ℃和250 ℃時(shí)分別在2.6 min和6.3 min內(nèi)釋放出6.2%(質(zhì)量分?jǐn)?shù))H2。此外,Nb2O5顆粒均勻分布在MgH2基體表面,并與MOF通過協(xié)同催化作用提高了MgH2的儲(chǔ)氫性能。
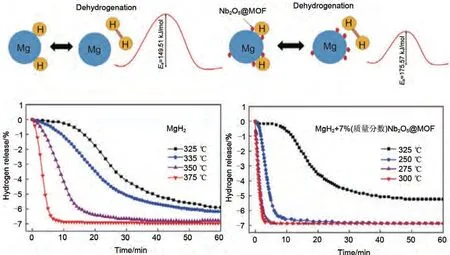
圖37 Nb2O5和MOF協(xié)同改善MgH2的脫氫性能機(jī)理圖和脫氫曲線[157]Fig.37 The mechanism diagram and corresponding dehydrogenation curve of MgH2 improved by Nb2O5 and MOF [157]
總體來說,MOF 自身可作為催化劑,也可作為其他催化劑或者M(jìn)g/MgH2的成核位點(diǎn)用于改善儲(chǔ)氫材料的動(dòng)力學(xué)/熱力學(xué)性能。此外,相比過渡金屬單質(zhì)的簡單添加,以過渡金屬為中心原子的MOF 材料具有更為優(yōu)異的結(jié)構(gòu),吸脫氫性能也有了進(jìn)一步改善。部分MOF改性的MgH2的儲(chǔ)氫性能匯總見表6。
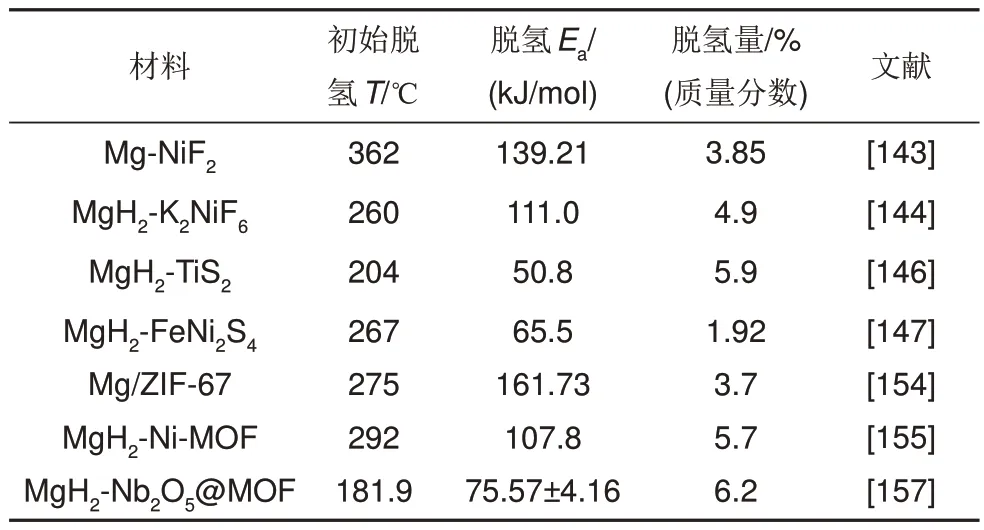
表6 部分其他金屬化合物改性的MgH2的儲(chǔ)氫性能匯總表Table 6 Summary of hydrogen storage properties of MgH2 modified by some other metal compounds
2.4 金屬與碳基復(fù)合催化劑改性
催化劑的效果不僅與其本身性質(zhì)有關(guān),其形態(tài)、粒徑、分散性等因素也顯著影響其催化活性。碳基材料是一種良好的催化劑載體,更重要的是碳基材料可以有效抑制MgH2顆粒的團(tuán)聚和長大。碳基材料(碳納米管、石墨烯、MXene等)具有良好的導(dǎo)電性、化學(xué)穩(wěn)定性,較高的比表面積等一系列優(yōu)點(diǎn),廣泛應(yīng)用于催化和儲(chǔ)氫領(lǐng)域。與其他儲(chǔ)氫材料相比,碳基材料具有較小的質(zhì)量密度,有利于提升整個(gè)體系的質(zhì)量儲(chǔ)氫密度,因此受到了研究者越來越多的關(guān)注[158-159]。
2.4.1 碳納米管
碳納米管(carbon nanotube, CNT)是碳材料家族的一位新成員,系Iijima[160]于1991 年首次發(fā)現(xiàn)。CNT是將單層石墨烯卷曲閉合,呈六邊形排列的碳原子構(gòu)成圓管,單壁碳納米管(SWCNT)只有一層,多壁碳納米管(MWCNT)為數(shù)層的同軸層狀中空結(jié)構(gòu)圓管,具有重量輕、化學(xué)穩(wěn)定性高、機(jī)械強(qiáng)度高等特點(diǎn),CNT層間可包覆MgH2/Mg防止團(tuán)聚現(xiàn)象,而且納米材料所具有的量子效應(yīng)、表面效應(yīng)等特性使其對氫氣吸附具有增強(qiáng)效應(yīng)。
繼Iijima 首次發(fā)現(xiàn)CNT 后,研究人員對CNT在儲(chǔ)氫方面的應(yīng)用進(jìn)行了各種探索。Kajiura 等[161]發(fā)現(xiàn)SWCNT、MWCNT 在環(huán)境溫度和高達(dá)8 MPa的壓力下儲(chǔ)氫量沒有超過0.43%(質(zhì)量分?jǐn)?shù));Ritschel等[162]對不同碳納米結(jié)構(gòu)的儲(chǔ)氫能力進(jìn)行研究發(fā)現(xiàn),純化的SWCNT在室溫和4.5 MPa的壓力下可逆儲(chǔ)氫容量僅為0.63%(質(zhì)量分?jǐn)?shù)),可以看出碳納米管需要在低溫高壓下才能具有一定的吸氫能力。Mosquera-Vargas 等[163]研究發(fā)現(xiàn),具有729.4 m2/g 比表面積的純化MWCNT 在室溫和12.79 kPa低壓條件下最大可以吸收3.46%(質(zhì)量分?jǐn)?shù))H2,但儲(chǔ)氫量仍不理想。近年來隨著MgH2高容量儲(chǔ)氫材料的快速發(fā)展,許多研究人員開始將擁有豐富成核位點(diǎn)的碳納米管作為金屬的優(yōu)良載體,用于改善MgH2的吸脫氫性能,取得了一系列成果。
Liu 等[164]將納米級雙向催化劑Co/Pd 均勻地分散在少壁的竹節(jié)狀碳納米管(B-CNTs)上,并通過球磨引入MgH2中。該復(fù)合材料對氫的吸附和解吸均表現(xiàn)出較好的催化效果。提出了如下的儲(chǔ)氫機(jī)理(圖38):①在球磨過程中,B-CNTs被機(jī)械力撕裂成少壁碳片。這些支撐Co/PdNPs的撕裂碳片覆蓋在MgH2顆粒表面,形成區(qū)隔層,防止了MgH2納米顆粒的團(tuán)聚和燒結(jié),提高了循環(huán)穩(wěn)定性。②B-CNTs具有大直徑(>100 nm)和高比表面積(1468 m2/g),促進(jìn)了Co/PdNPs 的均勻分散,增加了自身與MgH2納米顆粒的接觸面積,為氫氣擴(kuò)散提供了新的通道。③在氫化過程中,元素Pd起著主要的加速作用。氫原子在Pd/Mg界面優(yōu)先擴(kuò)散。而在脫氫過程中,Mg2Co與Mg2CoH5以及Mg Pd合金之間的相變降低了擴(kuò)散勢壘從而促進(jìn)氫原子釋放。MgH2-Co/Pd@B-CNTs 復(fù)合材料在198.9 ℃時(shí)開始脫氫,比研磨后的MgH2低132.4 ℃。在Co/Pd@B-CNTs的催化作用下,MgH2的脫氫活化能由178.0 kJ/mol降低到76.66 kJ/mol。同時(shí)該復(fù)合材料也表現(xiàn)出了優(yōu)異的動(dòng)力學(xué)性能,在250 ℃下10 s 內(nèi)快速吸附6.68%(質(zhì)量分?jǐn)?shù))H2,即使在50 ℃下100 s 內(nèi)也有1.91%(質(zhì)量分?jǐn)?shù))吸氫量,實(shí)現(xiàn)了吸脫氫量和熱/動(dòng)力學(xué)性能的雙重調(diào)控。
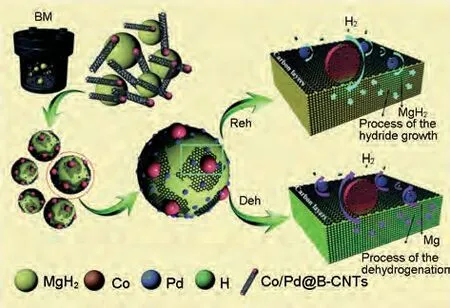
圖38 Co/Pd@B-CNTs對MgH2脫氫和吸氫的“雙向催化”機(jī)理示意圖[164]Fig.38 Schematic illustration for the “bidirectional catalysis” mechanism of Co/Pd@B-CNTs on the dehydrogenation and hydrogenation of MgH2[164]
Zhang 等[165]采用簡單的凝膠過濾和煅燒法制備了Ni@CNT 和多孔Ni3ZnC0.7顆粒的復(fù)合催化劑(Ni3ZnC0.7/Ni@CNT),并通過高效球磨引入高純MgH2中。如圖39 所示,MgH2-5%(質(zhì)量分?jǐn)?shù))Ni3ZnC0.7/Ni@CNT復(fù)合材料經(jīng)過第一次氫解吸后,會(huì)原位分解為Mg2Ni 和Zn。Zn/MgZn2的可逆相變以及Mg2Ni/Mg2NiH4的“氫泵”效應(yīng)能夠在氫吸附和解吸過程中為氫提供更多的成核位點(diǎn)和擴(kuò)散通道。Ni@CNT的存在也可以有效抑制納米復(fù)合材料在吸脫氫循環(huán)過程中的團(tuán)聚和燒結(jié)。該材料在80 ℃下60 min內(nèi)能吸附2.34%(質(zhì)量分?jǐn)?shù))H2,在300 ℃時(shí)釋放約5.36%(質(zhì)量分?jǐn)?shù))H2。MgH2-5%(質(zhì)量分?jǐn)?shù))Ni3ZnC0.7/Ni@CNT復(fù)合材料的吸氫活化能和脫氫活化能分別降至37.28 kJ/mol 和84.22 kJ/mol[圖40(a)、(b)]。此外,MgH2-5%(質(zhì)量分?jǐn)?shù))Ni3ZnC0.7/Ni@CNT復(fù)合材料的脫氫起始溫度降至約110 ℃,而純MgH2在約400 ℃高溫下才能開始脫氫[圖40(c)]。
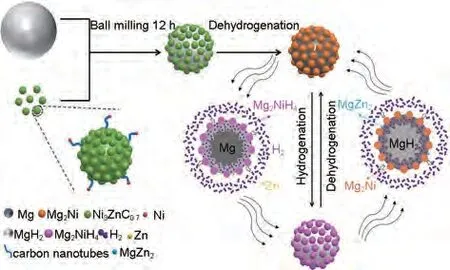
圖39 MgH2-5%(質(zhì)量分?jǐn)?shù))Ni3ZnC0.7/Ni@CNT納米復(fù)合材料吸/脫氫過程中催化劑催化機(jī)理示意圖[165]Fig.39 Schematic diagram of the catalytic mechanism of catalysts during the hydrogenation/dehydrogenation processes of the MgH2-5 wt%Ni3ZnC0.7/Ni@CNT nanocomposite[165]
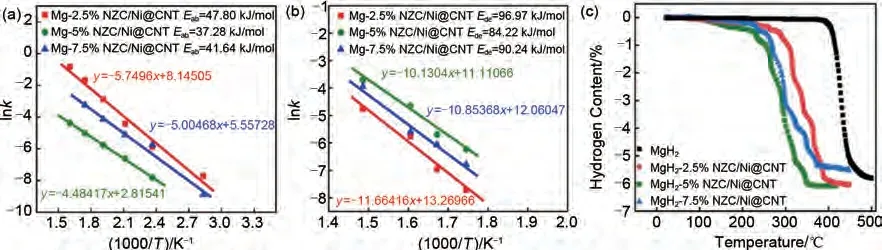
圖40 氫化態(tài)(a)和脫氫態(tài)(b) MgH2-xNZC/Ni@CNT(x=2.5%,5%,7.5%,質(zhì)量分?jǐn)?shù))復(fù)合材料的相應(yīng)lnk與1000/T圖;(c) MgH2-xNZC/Ni@CNT(x=2.5%,5%,7.5%,質(zhì)量分?jǐn)?shù))和商用MgH2的TPD曲線[165]Fig.40 The corresponding lnk vs 1000/T plots for hydrogenated (a) and dehydrogenated (b) MgH2-xNZC/Ni@CNT (x=2.5%, 5%, 7.5%)composites.Comparison of TPD curves (c) of MgH2-xNZC/Ni@CNT (x=2.5%, 5%,7.5%)and MgH2[165]
可見,碳納米管可制備成比表面積較大的竹節(jié)狀等形貌從而作為過渡金屬催化劑的高效載體用于改善鎂基儲(chǔ)氫材料的儲(chǔ)氫性能。
2.4.2 石墨烯
石墨烯(Graphene)是通過碳原子的sp2雜化緊密堆積所形成的具有二維六元環(huán)單層結(jié)構(gòu)的碳材料。自2004年曼徹斯特大學(xué)制備出能夠在室溫下穩(wěn)定存在的石墨烯薄片材料后,其研究得到了快速的發(fā)展。2015年,Zhang等[166]采用實(shí)驗(yàn)和第一性原理計(jì)算的方法,以晶格組成為Mg4H8的物理模型首次系統(tǒng)研究了Graphene 對MgH2脫氫性能的催化作用和機(jī)理。研究發(fā)現(xiàn)Graphene納米片分散包覆在MgH2顆粒外表面,有效抑制了球磨過程中MgH2顆粒的團(tuán)聚。石墨烯改性的機(jī)理在于同時(shí)降低了MgH2的脫氫焓和脫氫活化能,活化能從169.25 kJ/mol 降至90.01 kJ/mol(圖41)。
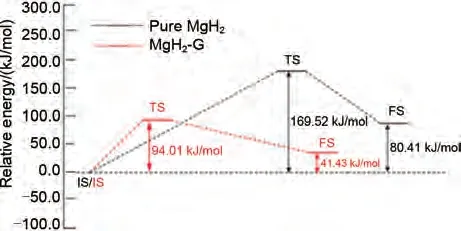
圖41 Mg4H8放氫過程的焓變 (a) 和石墨烯引入MgH2的活化能 (b)[166]Fig.41 (a) enthalpy change of graphene-doped Mg4H8 dehydrogenation and (b) activation energy of MgH2[166]
與碳納米管類似,石墨烯也具有較高的比表面積,可以為其他過渡金屬等催化劑提供豐富的負(fù)載位點(diǎn),從而協(xié)同改善MgH2的吸脫氫性能。陳萍團(tuán)隊(duì)[167]通過真空煅燒和機(jī)械球磨的方法成功制備了MgH2+10%(質(zhì)量分?jǐn)?shù))Fe-Ni@3DG(三維石墨烯)復(fù)合材料。如圖42(a)、(b)所示,MgH2+10%(質(zhì)量分?jǐn)?shù))Fe-Ni@3DG 復(fù)合體系可以在300 ℃下100 s 內(nèi)吸收6.35%(質(zhì)量分?jǐn)?shù))H2,并在500 s 內(nèi)釋放5.13%(質(zhì)量分?jǐn)?shù))H2。此外,在7 次循環(huán)吸脫氫性能測試中該體系能夠在10分鐘內(nèi)吸收6.5%(質(zhì)量分?jǐn)?shù))H2并釋放5.7%(質(zhì)量分?jǐn)?shù))H2[圖42(d)],明顯優(yōu)于球磨態(tài)MgH2的循環(huán)性能[圖42(c)],表現(xiàn)出優(yōu)異的循環(huán)穩(wěn)定性。兩種過渡金屬和石墨烯的多重催化作用進(jìn)一步改善了MgH2的吸脫氫性能。
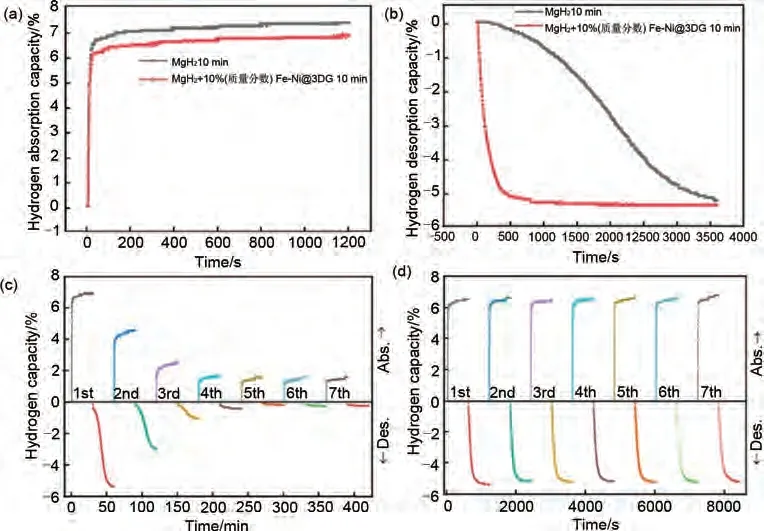
圖42 MgH2與MgH2+10%(質(zhì)量分?jǐn)?shù))Fe-Ni@3DG吸脫氫曲線:(a) 等溫吸氫;(b) 等溫脫氫;(c) MgH2循環(huán)性能圖;(d) MgH2+10%(質(zhì)量分?jǐn)?shù))Fe-Ni@3DG循環(huán)性能圖[167]Fig.42 Adsorption dehydrogenation curves of MgH2 and MgH2+10%Fe-Ni@3DG: (a) isothermal hydrogen absorption; (b) isothermal dehydrogenation; (c) cyclic performance diagram of MgH2; (d) cyclic performance diagram of MgH2+10%Fe-Ni@3DG[167]
因此,石墨烯自身可作為催化劑改善儲(chǔ)氫材料的活化能、焓變等參數(shù),也可作為其他催化劑的載體通過協(xié)同催化作用改善儲(chǔ)氫材料的吸脫氫循環(huán)性能。
2.4.3 金屬碳化物/氮化物
二維過渡金屬碳化物/氮化物(MXene)是一種具有層狀結(jié)構(gòu)[114]的新興金屬碳/氮基材料,具有化學(xué)耐久性、導(dǎo)熱性能好、力學(xué)性能好等優(yōu)點(diǎn),在提高M(jìn)gH2儲(chǔ)氫性能方面顯示出較高的催化活性[168-170]。對于二維材料MXene Ti3C2的研究開始于2011年[171],隨后在MXene 的合成、性能和多用途應(yīng)用等方面的研究開始不斷深入。MXene 的通式為MnXTx,其中M為部分過渡金屬,如Ti、Mo、Nb、V、Cr、Zr、Ta 等,n=1~4,X 為C 或N,Tx 為表面基團(tuán)。過渡金屬原子與碳原子以交替分層方式排列的結(jié)構(gòu)配置,使MXene具有很高的成分多樣性。
然而,與其他二維材料類似,由于表面終端基團(tuán)(—OH、—O、F)和范德華相互作用引起的層間耦合傾向,MXene 薄片在干燥過程中容易重新堆疊,導(dǎo)致用于錨定MgH2/Mg 材料的自由表面產(chǎn)生巨大損失[172]。此外,MXene 中大量的含氧端(—OH、—O)會(huì)與MgH2/Mg 納米顆粒反應(yīng)生成MgO或Mg(OH)2,導(dǎo)致負(fù)載量和脫氫動(dòng)力學(xué)降低。
為克服以上問題,Zhu等[173]利用具有折疊納米片形態(tài)的分層Ti3C2Tx 作為支撐材料,錨定了超分散的MgH2/Mg 納米顆粒。通過分散的Ti3C2Tx 納米片與十六烷基三甲基溴化銨(CTAB)分子之間的靜電相互作用制備了3D結(jié)構(gòu)的Ti3C2Tx。隨后,通過前驅(qū)體MgBu2自下向上自組裝的方法在煅燒Ti3C2薄片(Ti-MX)表面原位合成了超分散的MgH2NPs。得到的MgH2@Ti-MX 表現(xiàn)出快速的脫氫動(dòng)力學(xué),并且具有優(yōu)異的結(jié)構(gòu)和循環(huán)穩(wěn)定性。由圖43(a)~(c)可以看出,含有60%(質(zhì)量分?jǐn)?shù))MgH2NPs 的復(fù)合材料在140 ℃時(shí)開始脫氫,在150 ℃時(shí)2.5 h 內(nèi)能夠釋放3.0%(質(zhì)量分?jǐn)?shù))H2。此外,在200 ℃下循環(huán)60 次后仍然保持了高達(dá)4.0%(質(zhì)量分?jǐn)?shù))H2的可逆儲(chǔ)氫容量,動(dòng)力學(xué)方面沒有明顯損失。納米束縛和MgH2/Mg與Ti-MX之間的多相界面所引起的納米尺寸效應(yīng),特別是原位形成的催化相TiH2,是該材料優(yōu)越的吸氫性能的主要原因。
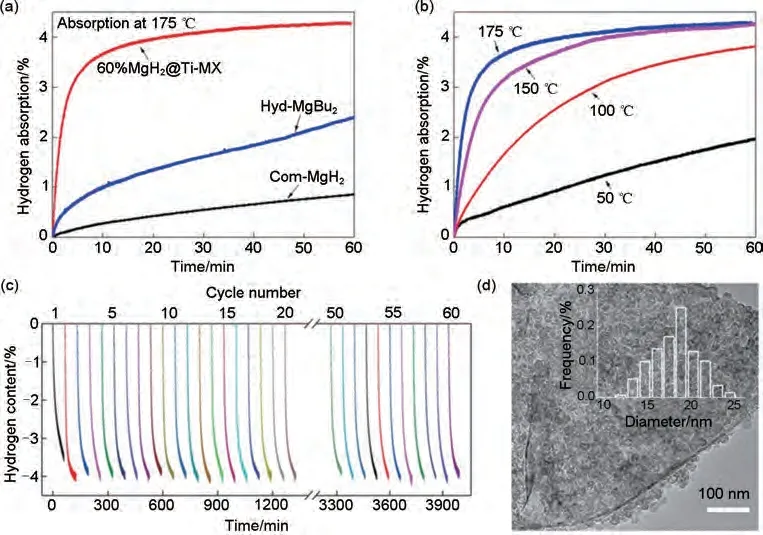
圖43 (a)商用MgH2、hyd-MgBu2和60%(質(zhì)量分?jǐn)?shù))MgH2@Ti-MX在175 ℃的等溫吸氫曲線; (b) 60%(質(zhì)量分?jǐn)?shù))MgH2@Ti-MX在不同溫度下的等溫吸氫曲線; (c) 60%(質(zhì)量分?jǐn)?shù))MgH2@Ti-MX在200℃下的循環(huán)脫氫曲線;(d) 60%(質(zhì)量分?jǐn)?shù))MgH2@Ti-MX在200℃下60次脫氫循環(huán)后的典型TEM圖像[173]Fig.43 (a) Isothermal hydrogenation curves of commercial MgH2, hyd-MgBu2, and 60% MgH2@Ti-MX at 175 ℃and (b) isothermal hydrogenation curves of 60% MgH2@Ti-MX at different temperatures; (c) cycling dehydrogenation curves of 60% MgH2@Ti-MX at 200 ℃, and (d) typical TEM image of 60% MgH2@Ti-MX after 60 de/hydrogenation cycles at 200 ℃[173]
Liu 等[174]通 過 剝 離 法 制 備 了V2C MXene 與Ti3C2MXene,之后將其與MgH2進(jìn)行球磨制備了MgH2-2V2C/Ti3C2復(fù)合材料。加入10%(質(zhì)量分?jǐn)?shù))2V2C/Ti3C2的MgH2在180 ℃左右開始脫氫,在225 ℃下60 min 內(nèi)可脫附5.1%(質(zhì)量分?jǐn)?shù))H2,并且在40 ℃室溫條件下20 s 內(nèi)可吸附5.1%(質(zhì)量分?jǐn)?shù))H2。6.3%(質(zhì)量分?jǐn)?shù))的可逆儲(chǔ)氫量在10 次循環(huán)中沒有明顯下降,表現(xiàn)出了優(yōu)異的循環(huán)穩(wěn)定性。圖44(a)表明在解吸過程中,氫原子或分子優(yōu)先通過MgH2/V2C/Ti3C2三晶界轉(zhuǎn)移,在吸附過程中,氫原子或分子優(yōu)先通過Mg/Ti3C2界面轉(zhuǎn)移。圖44(b)顯示了純MgH2和MgH2-2V2C/Ti3C2體系分解所需的能量。純MgH2的脫氫活化能為127 kJ/mol,反應(yīng)焓為78 kJ/mol,需要跳過49 kJ/mol勢壘。對于MgH2-2V2C/Ti3C2體系,脫氫活化能降至79 kJ/mol,反應(yīng)焓為76 kJ/mol,跳過勢壘僅為3 kJ/mol。復(fù)合材料較低的活化能以及V2C/Ti3C2對MgH2的輕微失穩(wěn)效應(yīng)共同促進(jìn)了MgH2儲(chǔ)氫性能的提高。
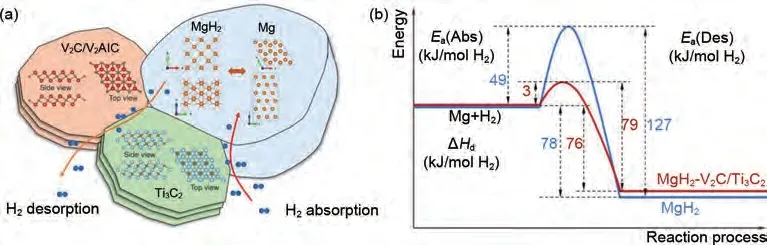
圖44 (a) 加入V2C/Ti3C2后MgH2的氫解吸機(jī)理示意圖;(b) MgH2和MgH2-V2C/Ti3C2的能壘對比示意圖[174]Fig.44 (a) Schematic pictures showing the hydrogen desorption and absorption mechanisms of MgH2 with addition of V2C/Ti3C2 and (b) schematic diagram comparatively displaying the energy barriers for as-milled MgH2 and MgH2-V2C/Ti3C2[174]
可見,具有多層結(jié)構(gòu)的MXene材料引入MgH2中可以為氫的吸附和解吸提供豐富的成核位點(diǎn)與擴(kuò)散通道,將其與一些過渡金屬復(fù)合,也可以有效改善MgH2的熱力學(xué)/動(dòng)力學(xué)性能。
綜上所述,碳材料可以有效地改善鎂基儲(chǔ)氫材料的儲(chǔ)氫性能,一方面可以為Mg/MgH2提供大量的新鮮表面并對其進(jìn)行包覆,既減小了顆粒尺寸又起到了抑制團(tuán)聚的作用;另一方面可以將催化劑負(fù)載到碳材料上,既可以減小催化劑的顆粒大小,也可以使催化劑更好地分散到鎂基復(fù)合材料中。因此,在改善鎂基復(fù)合材料的儲(chǔ)氫性能方面,碳材料起到至關(guān)重要的作用。
部分金屬與碳基復(fù)合催化劑改性的MgH2的儲(chǔ)氫性能如表7所示。
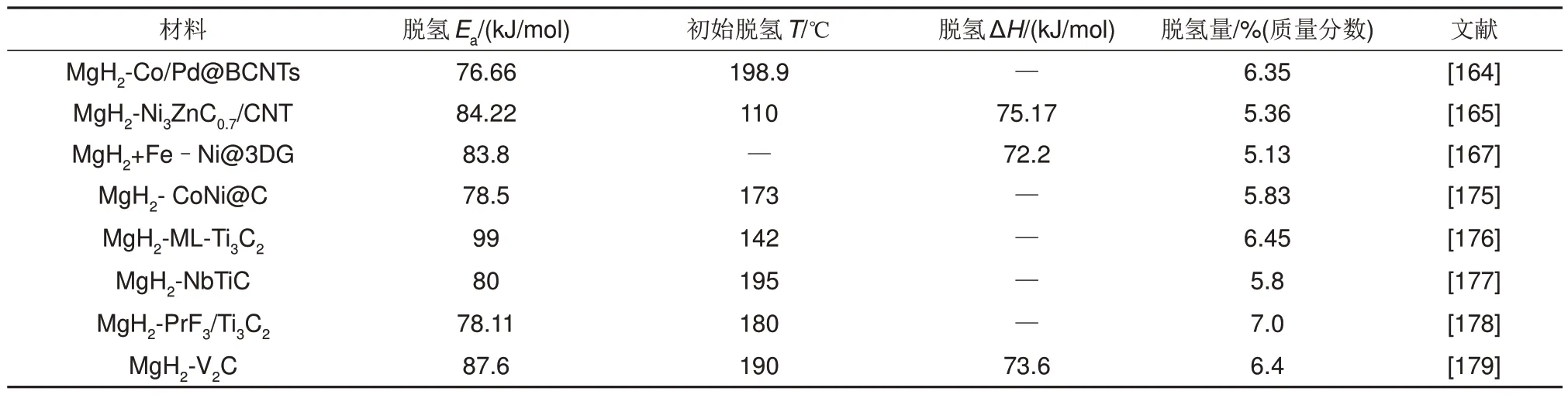
表7 部分金屬與碳基復(fù)合催化劑改性的MgH2的儲(chǔ)氫性能匯總表Table 7 Hydrogen storage performance of MgH2 modified by some metal and carbon based composite catalysts
2.5 高熵合金催化劑改性
合金化改性方面,鎂基HEA 受到了廣泛的研究,但由于多個(gè)金屬的引入,合金儲(chǔ)氫量發(fā)生了明顯下降。為了解決此問題,學(xué)者們提出了將不含鎂的HEA 直接引入MgH2中,由于MgH2本身就是具有可觀儲(chǔ)氫量的飽和吸氫態(tài),從而使MgH2-HEA復(fù)合體系兼顧了理想的吸脫氫量和多個(gè)金屬元素的多重催化效應(yīng)。近年來,得到了儲(chǔ)氫領(lǐng)域相關(guān)科研工作者的深入研究。
潘復(fù)生團(tuán)隊(duì)[180]采用熔煉- 霧化法制備FeCoNiCrMn高熵合金(HEA),通過球磨引入MgH2中成功制備了MgH2-5%(質(zhì)量分?jǐn)?shù))FeCoNiCrMn 復(fù)合儲(chǔ)氫體系。圖45(a)分別顯示了MgH2和MgH2-5%(質(zhì)量分?jǐn)?shù))FeCoNiCrMn 的TPD曲線。可以清楚地看到,HEA使MgH2的初始脫氫溫度(278.2 ℃)降低至209.1 ℃。吸脫氫量方面,圖45(b)表明MgH2-5%(質(zhì)量分?jǐn)?shù))FeCoNiCrMn 復(fù)合材料在250 ℃、300 ℃和350 ℃下脫氫量分別為0.72%、3.76%和5.98%。在相同條件下,研磨的MgH2的脫氫量為0、0.29%和5.21%,在450 ℃時(shí),MgH2-5%(質(zhì)量分?jǐn)?shù))FeCoNiCrMn 的脫氫量達(dá)到6.1%(質(zhì)量分?jǐn)?shù)),說明HEA的加入對儲(chǔ)氫能力的抑制并不嚴(yán)重。圖45(c)顯示了MgH2-5%(質(zhì)量分?jǐn)?shù))FeCoNiCrMn體系在260 ℃、280 ℃、300 ℃和325 ℃溫度下的等溫脫氫曲線。可以看出,在260 ℃下30 分鐘內(nèi)脫附了4.8%(質(zhì)量分?jǐn)?shù))H2。而研磨態(tài)MgH2即使在高達(dá)300 ℃條件下也幾乎不脫氫[圖45(e)]。圖45(d)顯示了MgH2-5%(質(zhì)量分?jǐn)?shù))FeCoNiCrMn 體系在50 ℃、100 ℃、150 ℃和250 ℃溫度下的等溫吸氫曲線。在250 ℃下22 min 內(nèi)的吸氫量達(dá)到6.0%(質(zhì)量分?jǐn)?shù)),而在相同溫度條件下,MgH2的吸氫量僅有4.0%(質(zhì)量分?jǐn)?shù))[見圖45(f)]。MgH2-5%(質(zhì)量分?jǐn)?shù))FeCoNiCrMn 一系列優(yōu)異性能證明了MgH2-高熵合金體系相比含鎂高熵合金體系更加可觀的儲(chǔ)氫量。
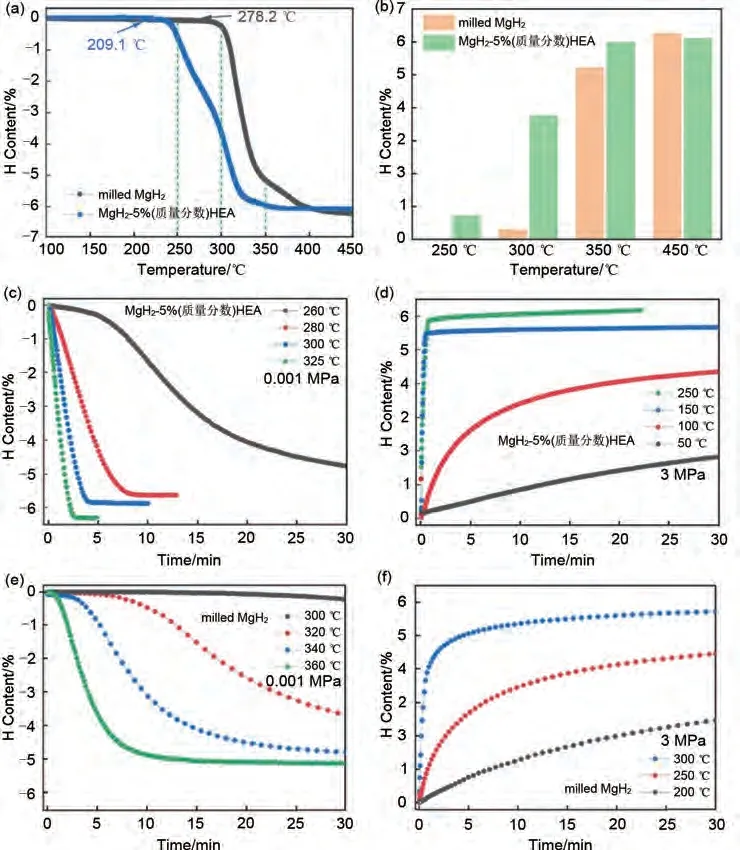
圖45 MgH2-5%(質(zhì)量分?jǐn)?shù))FeCoNiCrMn和研磨態(tài)MgH2的性能比較[180]:(a)TPD曲線;(b)加熱到不同溫度時(shí)的脫氫量;(c)、(e)等溫脫氫曲線;(d)、(f)等溫吸氫曲線Fig.45 Comparison of MgH2-5%FeCoNiCrMn and milled MgH2[180]: (a) TPD curves; (b) dehydrogenation content when heated to different temperatures; (c)、(e) isothermal dehydrogenation curves ; (d)、(f) isothermal hydrogenation curves
Zhang 等[181]制備了TiVNbZrFe、TiVNbZrNi 和TiVNbCrNi 用于改善 MgH2的儲(chǔ)氫性能,TiVNbZrFe 催化效果最佳。MgH2-TiVNbZrFe 體系在大約209 ℃開始釋放氫氣,比純MgH2低近170 ℃,在100 次循環(huán)內(nèi)具有高達(dá)6.16%(質(zhì)量分?jǐn)?shù))的可逆儲(chǔ)氫量,脫氫活化能降至63.03 kJ/mol。Verma 等[182]通過氫氧化鈉浸泡制備了MgH2-Al20Cr16Mn16Fe16Co16Ni16(LHEA),相比未浸泡MgH2-Al20Cr16Mn16Fe16Co16Ni16(HEA), MgH2-Al20Cr16Mn16Fe16Co16Ni16(LHEA)的初始脫氫溫度及最大脫氫量由MgH2-Al20Cr16Mn16Fe16Co16Ni16(HEA)的345 ℃進(jìn)一步降至338 ℃,最大脫氫量相比HEA 催化體系(6.3%,質(zhì)量分?jǐn)?shù))增加至6.8%(質(zhì)量分?jǐn)?shù))。Cr、Mn、Fe、Co、Ni的協(xié)同催化所產(chǎn)生的“雞尾酒效應(yīng)”達(dá)到增強(qiáng)催化效果的目的。此外,由于多種元素隨機(jī)排列,表面非常不均勻,為MgH2脫氫和再氫化過程中氫的擴(kuò)散提供了更豐富的活性位點(diǎn)。具體催化機(jī)理如圖46所示。
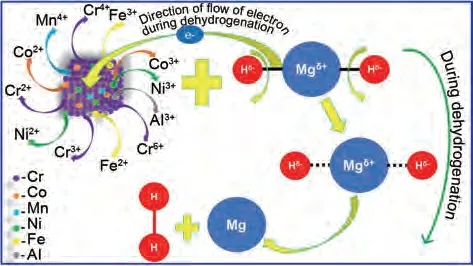
圖46 MgH2-LHEA體系的催化機(jī)理圖[181]Fig.46 Schematic representation of catalytic mechanism of catalyst (LHEA) during dehydrogenation of MgH2[181]
可以看出,MgH2-HEA復(fù)合體系相比含鎂HEA展現(xiàn)出更為優(yōu)異的吸脫氫性能,但HEA 組成元素的選擇與合金熔煉條件的優(yōu)化需要進(jìn)一步深入探索。
3 總結(jié)與展望
以青海察爾汗鹽湖、茶卡鹽湖為代表的鹵水鹽湖擁有豐富的鎂資源,保有儲(chǔ)量高達(dá)52.29 億噸,占全國已探明儲(chǔ)量的96.7%。因此探索鎂元素的高值化利用對鹽湖產(chǎn)業(yè)高質(zhì)量發(fā)展具有重要意義。
鎂基儲(chǔ)氫材料儲(chǔ)氫容量高,價(jià)格低廉,是一類具有極大發(fā)展?jié)摿Φ逆V基功能材料。然而,鎂基儲(chǔ)氫材料的實(shí)際應(yīng)用仍然受到放氫溫度較高、動(dòng)力學(xué)較差的限制。學(xué)者們采用合金化、納米化、催化劑摻雜等方法來改善鎂基材料的吸放氫熱/動(dòng)力學(xué)性能,取得了卓有成效的研究成果。
(1)合金化通過改變鎂基材料反應(yīng)路徑,降低吸脫氫活化能和焓來改善動(dòng)力/熱力學(xué)性能。然而,目前的鎂基合金儲(chǔ)氫密度較低,需要進(jìn)一步提高體系的儲(chǔ)氫密度。
(2)納米化技術(shù)在調(diào)控MgH2的熱/動(dòng)力學(xué)和可逆穩(wěn)定性方面也起著重要作用。研究表明,兼具“納米限域”和催化活性的高孔隙率、輕質(zhì)框架材料是制備納米Mg 材料的關(guān)鍵影響因素。因此,高穩(wěn)定性的多孔MOF 及其衍生物是未來制備納米鎂基材料的理想載體。
(3)金屬氧化物、氟化物、硫化物等催化劑能夠顯著提升鎂基儲(chǔ)氫材料的吸放氫熱/動(dòng)力學(xué)性能。添加劑本身或在制備/吸放氫過程中形成的催化相、活性界面能夠促進(jìn)H 的分解/解離及H 的擴(kuò)散,從而顯著提升鎂基材料的動(dòng)力學(xué)性能。另外,金屬與碳基復(fù)合催化劑對鎂基材料的循環(huán)穩(wěn)定性也有顯著改善作用,可以作為一類理想的催化添加劑在未來進(jìn)行更為深入的研究。
猜你喜歡
中學(xué)生數(shù)理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
建材發(fā)展導(dǎo)向(2021年14期)2021-08-23 00:56:16
紡織科技進(jìn)展(2021年3期)2021-06-09 08:07:14
中學(xué)生數(shù)理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
纖維復(fù)合材料(2018年3期)2018-04-25 07:22:58
電子測試(2017年11期)2017-12-15 08:57:13
產(chǎn)品可靠性報(bào)告(2017年7期)2017-09-05 09:49:12
汽車觀察(2016年3期)2016-02-28 13:16:26
應(yīng)用化工(2014年10期)2014-08-16 13:11:29
