2-甲酰基-1,1′-聯(lián)萘手性醛催化劑的合成
2024-02-28 01:40:10王煜洋郭其祥
合成化學(xué) 2024年2期
廖 健, 王煜洋, 郭其祥
(西南大學(xué) 化學(xué)化工學(xué)院,重慶 400715)
近年來,手性醛催化劑在手性胺合成化學(xué)中起到了越來越重要的作用。例如,手性醛可催化烯丙基胺與羥胺的不對(duì)稱氫胺化反應(yīng)[1],氨甲基類化合物的不對(duì)稱ɑ-官能團(tuán)化反應(yīng)[2-9],以及ɑ-羰基羧酸的轉(zhuǎn)胺化反應(yīng)[10]。目前所報(bào)道的手性醛催化劑可分為3大類(圖1):一是BEAUCHEMIN課題組[1]所報(bào)道的手性脂肪醛催化劑1;二是GUO課題組所報(bào)道的含有聯(lián)萘骨架的軸手性醛催化劑2[2]、3[3];三是ZHAO課題組所報(bào)道的手性吡哆醛類催化劑4[10]、5[4]。其中,GUO課題組所報(bào)道的手性醛催化劑3在催化甘氨酸衍生物與不飽和共軛烯酮的加成環(huán)化[4],以及氨基酸酯與對(duì)亞甲基苯醌的1,6-共軛加成反應(yīng)[5]中均取得了極其優(yōu)異的結(jié)果。

圖1 已報(bào)道的手性醛催化劑Figure 1 Reported chiral aldehyde catalysts
目前已報(bào)道的合成手性醛催化劑3的路線如圖2所示[3]。該路線經(jīng)9步反應(yīng),以約10%的總收率得到手性醛催化劑3,總收率較低。其主要原因?yàn)?由11經(jīng)DIBAL-H還原合成12時(shí)收率僅為50%。且在該合成路線中,由于帶吸電子取代基的芳基格試劑制備比較困難,因此未能得到具有吸電子芳基取代的手性醛催化劑3,這在一定程度上限制了手性醛催化劑3的生成。因此,對(duì)手性醛催化劑3的合成路線進(jìn)行改進(jìn)十分必要。

圖2 已報(bào)道的手性醛催化劑3的合成路線Figure 2 Reported synthesis route of chiral aldehyde catalysts 3
基于此,為了發(fā)展一種能夠以較高收率獲得不同取代基取代的手性醛催化劑3,且適用于大量合成的合成方法。因此,根據(jù)相關(guān)文獻(xiàn)報(bào)道[3,11-15],重新設(shè)計(jì)了手性醛催化劑3的合成路線(圖3)。 (S)-BINOL與對(duì)甲苯磺酰氯以及三氟甲磺酸酐進(jìn)行磺酰化反應(yīng)[11]得到(S)-1,1′-聯(lián)萘-2′-((三氟甲基磺酰基)氧基)-2-((4-甲基苯基磺酰基)氧基)(13),13在醋酸鈀的催化下進(jìn)行插羰反應(yīng)[11],得到(S)-1,1′-聯(lián)萘-2′-((4-甲基苯基磺酰基)氧基)-2-乙氧羰基(14)。14發(fā)生鎳催化的Suzuki偶聯(lián)反應(yīng)[11-12]得到(S)-1,1′-聯(lián)萘-2′-芳基-2-乙氧羰基(15),15在Mg(TMP)2[3,13]作用下,與硼酸三甲酯反應(yīng),經(jīng)鹽酸水解得到(S)-(2-(乙氧羰基)-2′-芳基-[1,1′-聯(lián)萘]-3-硼酸(16)。16在雙氧水作用下得到(S)-1,1′-聯(lián)萘-2′-芳基-3-羥基-2-甲酸乙酯(17),17經(jīng)溴甲基甲基醚保護(hù)[14]得到(S)-1,1′-聯(lián)萘-2′-芳基-3-(甲氧基甲氧基)-2-甲酸乙酯(18)。18經(jīng)氫化鋁鋰還原得到(S)-1,1′-聯(lián)萘-2′-芳基-3-(甲氧基甲氧基)-2-甲醇(19),19發(fā)生戴斯馬丁氧化反應(yīng)得到(S)-1,1′-聯(lián)萘-2′-芳基-3-(甲氧基甲氧基)-2-甲醛(20)。20經(jīng)鹽酸作用脫除甲氧基甲基得到12,12經(jīng)N-溴化琥珀酰亞胺(NBS)溴化[15]得到手性醛催化劑3。
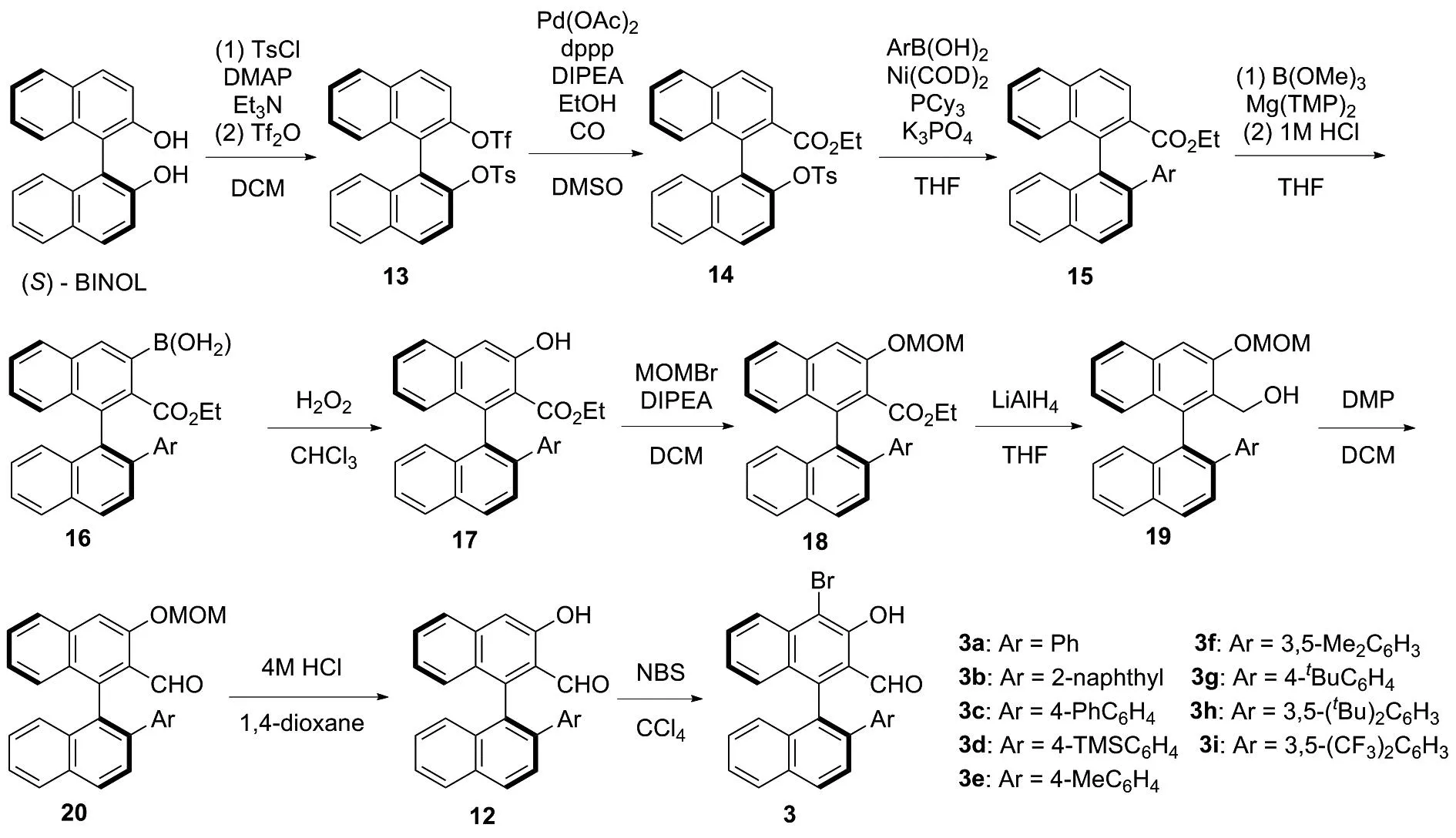
圖3 手性醛催化劑3的合成新路線Figure 3 New synthesis route of chiral aldehyde catalysts 3
1 實(shí)驗(yàn)部分
1.1 儀器與試劑
卓的-ZD90型電熱數(shù)字顯示熔點(diǎn)儀; Perkin-Elmer-341型自動(dòng)旋光儀; Bruker-400/600 MHz型核磁共振儀; Bruker-impact II型高分辨率質(zhì)譜儀。
所用試劑均為分析純。
1.2 合成
(1)13的合成
根據(jù)文獻(xiàn)[11]合成13。
(2)14的合成

(3)15a~15i的合成(以15b為例)
在手套箱中,于干燥的48 mL耐壓瓶中加入雙(1,5-環(huán)辛二烯)鎳(0.06 g, 0.23 mmol)、三環(huán)己基膦(0.25 g, 0.90 mmol)和四氫呋喃(5 mL),并用磁力攪拌子攪拌5 min。加入14(1.48 g, 3.00 mmol)、 2-萘硼酸(1.55 g, 9.00 mmol)、磷酸三鉀(1.91 g, 9.00 mmol)和四氫呋喃(10 mL),并于80 ℃下攪拌48 h。向反應(yīng)體系中加水稀釋,混合物用乙酸乙酯(3×15 mL)萃取,合并有機(jī)相。有機(jī)相經(jīng)無水硫酸鈉干燥后減壓濃縮,經(jīng)柱層析得15b(1.06 g)。經(jīng)由相同方法合成15a~15i。
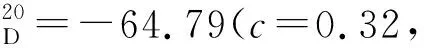





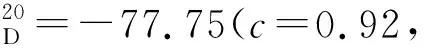


(4)16a~16i的合成(以16b為例)
在干燥的100 mL三頸燒瓶中加入15b(1.06 g, 2.30 mmol),并用磁力攪拌子攪拌,氮?dú)庵脫Q3次,加入四氫呋喃(10 mL)。將體系冷卻至-78 ℃,滴加Mg(TMP)2(26.00 mL, 11.50 mmol),攪拌5 min后升至室溫?cái)嚢? h。然后再將體系冷卻至-78 ℃,滴加硼酸三甲酯(1.50 mL, 13.80 mmol),于該溫度下攪拌30 min后升至室溫?cái)嚢?4 h。將體系冷卻至0 ℃,滴加15 mL 1M鹽酸,升至室溫繼續(xù)攪拌2 h。混合物用乙酸乙酯(3×15 mL)萃取,合并有機(jī)相。有機(jī)相經(jīng)無水硫酸鈉干燥后減壓濃縮得到16b,無需進(jìn)一步純化,直接進(jìn)行下一步投料。經(jīng)由相同方法合成16a~16i。
(5)17a~17i的合成(以17b為例)
在50 mL圓底燒瓶中加入16b、氯仿(10 mL),并用磁力攪拌子攪拌。將體系冷卻至0 ℃,滴加過氧化氫(質(zhì)量分?jǐn)?shù)為30%, 2.30 mL, 23.00 mmol),升至室溫?cái)嚢?0 h,再次將體系冷卻至0 ℃,加入飽和亞硫酸鈉溶液淬滅反應(yīng)。混合物用二氯甲烷(3×15 mL)萃取,合并有機(jī)相。有機(jī)相經(jīng)無水硫酸鈉干燥后減壓濃縮得到17b,無需進(jìn)一步純化,直接進(jìn)行下一步投料。經(jīng)由相同方法合成17a~17i。
(6)18a~18i的合成(以18b為例)
在50 mL圓底燒瓶中加入17b、N,N-二異丙基乙胺(1.60 mL, 9.20 mmol),并用磁力攪拌子攪拌,加入二氯甲烷(10 mL),將體系冷卻至0 ℃,滴加溴甲基甲基醚(0.30 mL, 3.45 mmol)。升至室溫?cái)嚢? h,再次將體系冷卻至0 ℃,加水淬滅反應(yīng)。混合物用二氯甲烷(3×15 mL)萃取,合并有機(jī)相。有機(jī)相經(jīng)無水硫酸鈉干燥后減壓濃縮,經(jīng)柱層析得18b(0.85 g)。經(jīng)由相同方法合成18a~18i。
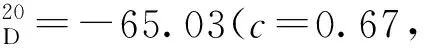


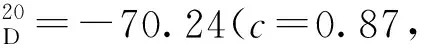



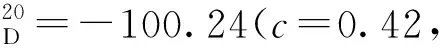
(7)19a~19i的合成(以19b為例)
在100 mL圓底燒瓶中加入18b(1.85 g, 1.67 mmol),并用磁力攪拌子攪拌。加入四氫呋喃(15 mL),將體系冷卻至0 ℃,緩慢加入氫化鋁鋰(0.13 g, 3.34 mmol)。升至室溫?cái)嚢? h,再次將體系冷卻至0 ℃,加水淬滅反應(yīng)。混合物用乙酸乙酯(3×15 mL)萃取,合并有機(jī)相。有機(jī)相經(jīng)無水硫酸鈉干燥后減壓濃縮得到19b,無需進(jìn)一步純化,直接進(jìn)行下一步投料。經(jīng)由相同方法合成19a~19i。
(8)20a~20i的合成(以20b為例)
在100 mL圓底燒瓶中加入19b,并用磁力攪拌子攪拌,加入二氯甲烷(10 mL),將體系冷卻至0 ℃,緩慢加入戴斯-馬丁氧化劑(DMP)(2.12 g, 5.01 mmol)。升至室溫?cái)嚢? h,再次將體系冷卻至0 ℃,加入飽和亞硫酸鈉溶液淬滅反應(yīng)。混合物用二氯甲烷(3×15 mL)萃取,合并有機(jī)相。有機(jī)相經(jīng)無水硫酸鈉干燥后減壓濃縮得到20b,無需進(jìn)一步純化,直接進(jìn)行下一步投料。經(jīng)由相同方法合成20a~20i。
(9)12a~12i的合成(以12b為例)
在50 mL圓底燒瓶中加入20b,并用磁力攪拌子攪拌,加入1,4-二氧六環(huán)(15 mL),將體系冷卻至0 ℃,加入4 M鹽酸(0.90 mL, 3.34 mmol)。 60 ℃下攪拌2 h,經(jīng)減壓濃縮后加水稀釋,混合物用乙酸乙酯(3×15 mL)萃取,合并有機(jī)相。有機(jī)相經(jīng)無水硫酸鈉干燥后減壓濃縮,經(jīng)柱層析得12b(0.60 g)。經(jīng)由相同方法合成12a~12i。
12a:黃色固體,收率83%;1H NMR(400 MHz, CDCl3)δ: 10.84(s, 1H), 9.47(s, 1H), 8.02(d,J=8.0 Hz, 1H), 7.91(d,J=8.0 Hz, 1H), 7.64~7.56(m, 2H), 7.44~7.37(m, 2H), 7.27~7.23(m, 2H), 7.20~7.16(m, 1H), 7.08~6.93(m, 5H), 6.90~6.86(m, 2H);13C NMR(101 MHz, CDCl3)δ: 197.4, 156.4, 146.4, 140.9, 140.8, 138.0, 133.4, 132.4, 129.9, 129.8, 129.4, 128.5, 128.4, 128.1, 128.0, 127.9, 127.8, 127.3, 127.2, 127.0, 126.8, 126.3, 124.5, 119.9, 112.1。
12b:黃色固體,收率85%;1H NMR(400 MHz, CDCl3)δ: 10.76(s, 1H), 9.53(s, 1H), 8.06(d,J=8.0 Hz, 1H), 7.94(d,J=8.0 Hz, 1H), 7.70~7.65(m, 1H), 7.63~7.57(m, 2H), 7.48~7.38(m, 5H), 7.35~7.24(m, 4H), 7.18~7.14(m, 1H), 7.13~7.04(m, 2H), 6.98~6.93(m, 1H);13C NMR(101 MHz, CDCl3)δ: 197.3, 156.4, 146.4, 140.8, 138.4, 138.0, 133.5, 132.9, 132.5, 132.2, 130.2, 129.9, 129.5, 128.5, 128.2, 128.1, 128.0, 127.9, 127.8, 127.7, 127.5, 127.3, 127.0, 126.8, 126.4, 126.3, 126.2, 126.1, 124.6, 120.0, 112.2。

12d:黃色固體,收率74%;1H NMR(400 MHz, CDCl3)δ: 10.87(s, 1H), 9.47(s, 1H), 8.01(d,J=8.0 Hz, 1H), 7.90(d,J=8.0 Hz, 1H), 7.64(d,J=8.0 Hz, 1H), 7.57(d,J=8.0 Hz, 1H), 7.45~7.38(m, 2H), 7.28~7.19(m, 3H), 7.14~7.10(m, 2H), 7.09~7.03(m, 1H), 6.99(d,J=8.0 Hz, 1H), 6.88(d,J=8.0 Hz, 2H), 0.08(s, 9H);13C NMR(101 MHz, CDCl3)δ: 197.4, 156.5, 146.5, 141.1, 140.8, 139.3, 138.0, 133.5, 133.1, 132.4, 129.8, 129.7, 129.4, 128.6, 128.1, 128.0, 127.9, 127.8, 127.2, 127.0, 126.8, 126.3, 124.5, 119.9, 112.1, -1.2。

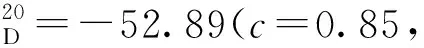
12g:黃色固體,收率79%;1H NMR(400 MHz, CDCl3)δ: 10.86(s, 1H), 9.44(d,J=8.0 Hz, 1H), 7.99(d,J=8.0 Hz, 1H), 7.88(d,J=8.0 Hz, 1H), 7.63(d,J=8.0 Hz, 1H), 7.57(d,J=12.0 Hz, 1H), 7.43~7.36(m, 2H), 7.27~7.15(m, 3H), 7.07~7.01(m, 1H), 7.00~6.95(m, 3H), 6.83~6.78(m, 2H), 1.10(s, 9H);13C NMR(101 MHz, CDCl3)δ: 197.5, 156.5, 150.0, 146.7, 140.8, 137.9, 137.7, 133.5, 132.3, 129.8, 129.7, 129.3, 128.6, 128.3, 128.2, 128.1, 128.0, 127.1, 127.0, 126.7, 126.1, 125.0, 124.5, 119.9, 112.0, 34.4, 31.1。


(10)3a~3i的合成(以3b為例)
在50 mL圓底燒瓶中加入12b(0.60 g, 1.40 mmol),并用磁力攪拌子攪拌。然后再加入四氯化碳(14 mL),將體系冷卻至0 ℃,緩慢加入NBS(0.37 g, 2.10 mmol),升至室溫?cái)嚢? h,再次將體系冷卻至0 ℃,滴加飽和亞硫酸鈉溶液淬滅反應(yīng)。混合物用二氯甲烷(3×15 mL)萃取,合并有機(jī)相。有機(jī)相經(jīng)無水硫酸鈉干燥后減壓濃縮,經(jīng)柱層析得3b(0.63 g)。經(jīng)由相同方法合成3a~3i。
3a:黃色固體,收率96%;1H NMR(400 MHz, CDCl3)δ: 11.72(s, 1H), 9.50(s, 1H), 8.26(d,J=8.0 Hz, 1H), 8.11(d,J=8.0 Hz, 1H), 7.99(d,J=8.0 Hz, 1H), 7.69~7.61(m, 2H), 7.51(t,J=8.0 Hz, 1H), 7.39(d,J=8.0 Hz, 1H), 7.34~7.30(m, 1H), 7.23~7.19(m, 1H), 7.11~7.03(m, 4H), 6.96~6.94(m, 2H);13C NMR(151 MHz, CDCl3)δ: 196.8, 153.3, 146.1, 141.1, 140.6, 136.1, 133.3, 132.4, 131.2, 129.7, 129.2, 129.1, 128.4, 128.3, 128.2, 128.1, 127.9, 127.5, 127.4, 126.6, 126.4, 126.3, 125.1, 119.9, 107.3。
3b:黃色固體,收率90%;1H NMR(400 MHz, CDCl3)δ: 11.65(s, 1H), 9.56(s, 1H), 8.25(d,J=8.0 Hz, 1H), 8.14(d,J=8.0 Hz, 1H), 8.01(d,J=8.0 Hz, 1H), 7.74(d,J=8.0 Hz, 1H), 7.68~7.62(m, 2H), 7.54~7.46(m, 5H), 7.39~7.32(m, 3H), 7.27~7.23(m 1H), 7.08(d,J=8.0 Hz, 1H), 7.02~6.99(m, 1H);13C NMR(151 MHz, CDCl3)δ: 196.8, 153.3, 146.1, 141.0, 138.2, 136.1, 133.4, 132.9, 132.5, 132.2, 131.3, 129.8, 129.4, 129.2, 128.4, 128.3, 128.1, 128.0, 127.9, 127.8, 127.6, 126.6, 126.5, 126.4, 126.3, 126.2, 126.1, 125.2, 119.9, 107.4。

3d:黃色固體,收率91%;1H NMR(600 MHz, CDCl3)δ: 11.59(s, 1H), 9.32(s, 1H), 8.10(d,J=6.0 Hz, 1H), 7.92(d,J=6.0 Hz, 1H), 7.80(d,J=6.0 Hz, 1H), 7.49~7.44(m, 2H), 7.35~7.30(m, 1H), 7.24(d,J=6.0 Hz, 1H), 7.13(t,J=6.0 Hz, 1H), 7.06~7.02(m, 3H), 6.84(d,J=6.0 Hz, 1H), 6.77(d,J=6.0 Hz, 2H), 0.00(s, 9H);13C NMR(151 MHz, CDCl3)δ: 196.9, 153.3, 146.2, 141.0, 140.8, 139.6, 136.1, 133.4, 133.2, 132.4, 131.3, 129.7, 129.3, 129.0, 128.5, 128.2, 128.0, 127.7, 127.4, 126.6, 126.4, 126.3, 125.1, 119.9, 107.3, -1.3。

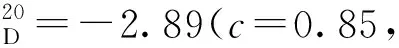
3g:黃色固體,收率92%;1H NMR(400 MHz, CDCl3)δ: 11.77(s, 1H), 9.48(s, 1H), 8.28(d,J=8.0 Hz, 1H), 8.09(d,J=8.0 Hz, 1H), 7.98(d,J=8.0 Hz, 1H), 7.69~7.61(m, 2H), 7.52~7.47(m, 1H), 7.43~7.38(m, 1H), 7.32~7.27(m, 1H), 7.24~7.19(m, 1H), 7.11~7.05(m, 2H), 7.03~6.98(m, 1H), 6.91~6.87(m, 2H), 1.20(s, 9H);13C NMR(101 MHz, CDCl3)δ: 197.0, 153.3, 150.2, 146.4, 140.9, 137.5, 136.0, 133.4, 132.3, 131.3, 129.6, 129.3, 128.9, 128.5, 128.2, 128.1, 127.4, 126.6, 126.4, 126.3, 125.2, 125.1, 119.8, 107.2, 34.4, 31.1。
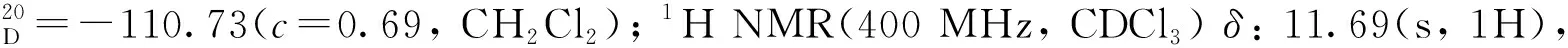

2 結(jié)果與討論
2.1 放大反應(yīng)
該合成方法中,由(S)-BINOL合成13,如圖4(a)所示,可在50 mmol的規(guī)模上得到90%的收率。13可以在20 mmol的規(guī)模上以71%的收率合成14,如圖4(b)所示。從14(10.00 mmol)出發(fā)合成3a,如圖4(c)所示,經(jīng)偶聯(lián)反應(yīng)、硼化、氧化、甲氧基甲基保護(hù)、還原、氧化、脫保護(hù)以及溴代8步反應(yīng)能以29%的總收率得到目標(biāo)產(chǎn)物。
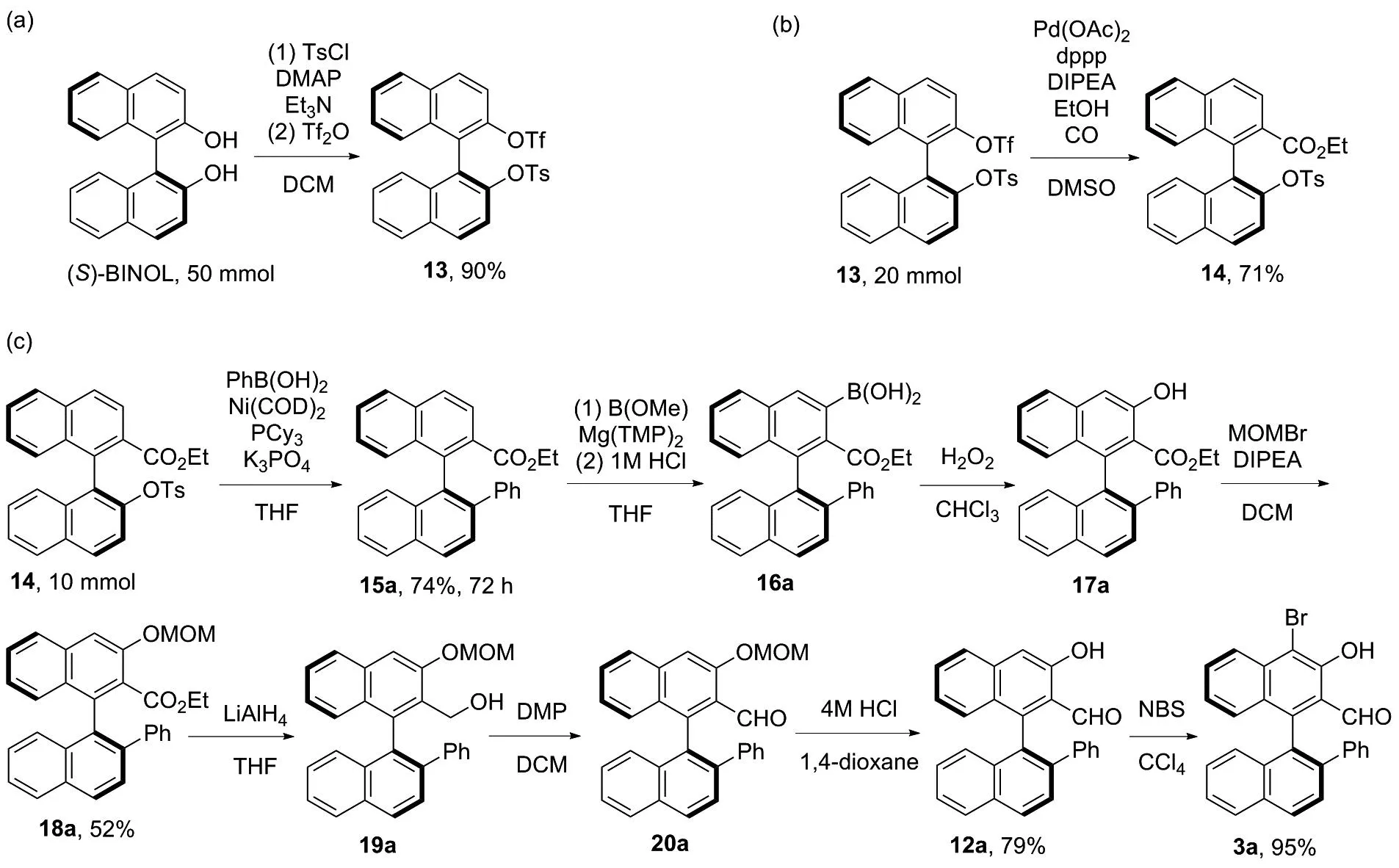
圖4 手性醛3a的克級(jí)規(guī)模合成Figure 4 Gram-scale synthesis for chiral aldehyde 3a
2.2 烷氧基對(duì)17合成的影響
在由13合成17的過程中,烷氧基對(duì)于17收率有較大的影響,因此在其它條件不變的情況下,考察了烷氧基對(duì)17收率的影響(圖5,表1)。當(dāng)烷氧基為甲氧基時(shí),有利于插羰反應(yīng)以及偶聯(lián)反應(yīng),能以29%的總收率得到目標(biāo)產(chǎn)物(Entry 1)。而使用正丙基氧基時(shí),則是有利于在酯基鄰位引入羥基,能以22%的總收率得到目標(biāo)產(chǎn)物(Entry 3)。當(dāng)選用乙氧基時(shí),各步反應(yīng)都能有中等的收率,能以36%的總收率得到目標(biāo)產(chǎn)物(Entry 2)。因此,乙氧基為最優(yōu)的烷氧基。

表1 烷氧基對(duì)17合成中收率的影響Table 1 Effect of alkoxy group on the synthesis of the yield of 17
2.3 17合成12的條件篩選
在由17合成12的過程中,對(duì)條件進(jìn)行了一系列篩選。首先選用DIBAL-H對(duì)17b直接進(jìn)行還原,能以36%收率得到12b,如圖6(a)所示。之后嘗試選用氫化鋁鋰對(duì)17b進(jìn)行還原,以63%收率得到(S)-1,1′-聯(lián)萘-2′-(2-萘基)-3-羥基-2-甲醇(21),再對(duì)21進(jìn)行Parikh-Doering氧化[16],能以43%收率進(jìn)行氧化,以總收率27%得到12b,如圖6(b)所示。此外,嘗試使用溴甲基甲基醚對(duì)17b酚羥基進(jìn)行保護(hù),能以90%收率得到18b,再使用氫化鋁鋰對(duì)18b進(jìn)行還原,能以83%收率得到19b。之后使用戴斯-馬丁氧化劑對(duì)19b進(jìn)行氧化,能以91%收率得到20b,使用鹽酸能以90%收率對(duì)保護(hù)基進(jìn)行脫除,以4步總收率61%得到12b,如圖6(c)所示。因此,選用收率最高的路徑,經(jīng)保護(hù)、還原、氧化和脫保護(hù)4步合成12b。
2.4 氧化劑的篩選
在合成20的過程中,發(fā)現(xiàn)氧化劑的種類會(huì)對(duì)反應(yīng)收率產(chǎn)生影響。因此在其它條件不變的情況下,考察了氧化劑類型對(duì)反應(yīng)的影響。在使用DMP時(shí),能以91%收率得到目標(biāo)產(chǎn)物(表2, Entry 1),相較于氯鉻酸吡啶鹽(PCC)效果更好(80%, Entry 2),因此選用DMP作氧化劑。

表2 氧化劑的篩選Table 2 Selection of oxidant
本文報(bào)道了手性醛催化劑(3)合成路線的優(yōu)化方法。相較于文獻(xiàn)中已報(bào)道的路線,該路線能以大于20%的總收率合成大部分2′-位具有更多類型取代基的3;能以12%的總收率合成在2′-位帶有強(qiáng)吸電子基團(tuán)的芳基(3,5-二(三氟甲基)苯基)的3,這對(duì)于多樣性3的獲取具有一定的積極作用。該合成路線避免了使用具有強(qiáng)腐蝕性的溴素作為溴源,而是選用對(duì)環(huán)境更加友好的NBS作為溴源進(jìn)行溴化反應(yīng),且該方法能放大到10 mmol規(guī)模來進(jìn)行,具有一定應(yīng)用前景。
猜你喜歡
新世紀(jì)智能(數(shù)學(xué)備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
浙江大學(xué)學(xué)報(bào)(工學(xué)版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
現(xiàn)代企業(yè)(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
應(yīng)用化工(2014年3期)2014-08-16 13:23:50
