超高效液相色譜-串聯質譜法檢測蘑菇中6種蘑菇毒素的不確定度評定
2024-02-20 09:12:38趙秀琳趙小林劉秋平彭珍華楊韻渲牛之瑞馬雪濤譚建林
化學分析計量 2024年1期
趙秀琳,趙小林,劉秋平,彭珍華,楊韻渲,牛之瑞,馬雪濤,譚建林
(云南省產品質量監督檢驗研究院,國家熱帶農副產品質量檢驗檢測中心,昆明 650223)
蘑菇為大型真菌的俗稱,大型真菌通常是指肉眼可見、徒手可摘的真菌[1]。按用途分為食用菌、藥用菌、有毒菌和用途未知菌四大類。蘑菇因其獨特的鮮味和滋味,受到越來越多的人青睞。特別是在每年雨季到來之際,野生食用菌更是食客必選的佳肴。野生菌種類繁多,單靠民間流傳的一些辨別方法,無法有效分辨其有無毒性,因此因食用野生蘑菇中毒的事件時有報道[2-5]。在毒菇中毒事故中,約90%是由于誤食含鵝膏毒肽和鬼筆毒肽的野生菌所引起[6]。蘑菇中毒病癥可分為急性肝損傷型、急性腎衰竭型、胃腸炎型、神經精神型、溶血型、光過敏性皮炎型六大類[7],臨床上常根據中毒癥狀和檢測患者尿液或血液中有無蘑菇毒素來判斷,從而進行相應治療。誤食毒菇中毒對患者身體的損傷是不可估量且難以挽回的,因此準確檢測和發現蘑菇中的毒素,幫助人們提前認識、區分毒蘑菇,從而減少誤食的情況,保護人類身體健康具有重要意義。
目前蘑菇毒素的檢測方法主要有毛細管電泳法[8]、酶聯免疫吸附法[9]、高效液相色譜法、液相色譜-串聯質譜法[10-11]和液相色譜-四級桿飛行時間質譜法[12-13],而在食品檢測標準中,蘑菇毒素的測定主要是液相色譜-串聯質譜法,該方法具有高效、快速、靈敏度高的特點,能夠在短時間內檢測大批量未知蘑菇樣品中蘑菇毒素的含量,因此在食品檢測中應用廣泛[14]。
測量不確定度表征賦予了被測量量值的分散性,是測量結果的一部分,也是符合性判定規則考慮的主要內容。檢測實驗室應分析測量不確定度對檢測結果的貢獻,評定每一項用數值表示的測量結果的測量不確定度。BJS 202008《蘑菇中α-鵝膏毒肽等6 種蘑菇毒素的測定》是食品行業檢測蘑菇毒素的專用標準方法,該方法測量不確定度的評定未見報道。筆者根據JJF 1059.1—2012《測量不確定度評定與表示》和CNAS-GL006《化學分析中的不確定度評估指南》,參照BJS 202008開展高效液相色譜-串聯質譜法檢測蘑菇中6種蘑菇毒素方法的不確定度評定,同時為測量其它藥物殘留量的不確定度分析提供參考。
1 實驗部分
1.1 主要儀器與試劑
超高效液相色譜-三重四級桿串聯質譜聯用儀:LC-MS 8050型,日本島津儀器有限公司。
電子天平:CPA225D 型,感量為0.1 mg,德國賽多利斯集團。
移液器:PETTE FIX 型,量程分別為200 μL 和1 000 μL,德國艾卡集團。
HLB 固相萃取小柱:60 mg/3 mL,美國安捷倫科技有限公司。
乙腈、甲醇、正己烷、乙酸銨:色譜純,上海安譜實驗科技股份有限公司。
α-鵝膏毒肽、β-鵝膏毒肽、γ-鵝膏毒肽、二羥基鬼筆毒肽、羧基二羥基鬼筆毒肽標準物質:質量分數分別為:97.3%、90.3%、95.0%、87.4%、98.4%,相對擴展不確定度均為2%(k=2),上海安譜實驗科技股份有限公司。
羧基三羥基鬼筆毒肽標準溶液:50 μg/mL,溶劑為水,相對擴展不確定度為2%(k=2),青島普瑞邦生物工程有限公司。
干香菇樣品:市售。
1.2 儀器工作條件
1.2.1 色譜條件
色譜柱:BEH-C18型色譜柱(50 mm×2.1 mm,1.7 μm,美國沃特世公司);柱溫:40 ℃;流動相:A 相為5 mmol/L 乙酸銨水溶液,B 相為甲醇;洗脫方式:梯度洗脫;洗脫程序:0~2.5 min時流動相B的體積分數為10%~20%,2.5~3.0 min 時流動相B 的體積分數為20%~45%,3.0~3.1 min 時流動相B 的體積分數為45%~95%,3.1~3.8 min 時流動相B 的體積分數為95%,3.8~3.85 min 時流動相B 的體積分數為95%~10%,3.85~6 min 時流動相B 的體積分數為10%;流量:0.3 mL/min;進樣體積:5 μL。
1.2.2 質譜條件
離子源:電噴霧正離子模式(ESI+);檢測方式:多反應監測(MRM)模式;接口電壓:4 000 V;霧化氣流量:2.9 L/min;加熱氣和干燥氣流量:10.0 L/min;接口溫度:300 ℃;脫溶劑管溫度:250 ℃;加熱塊溫度:400 ℃;碰撞氣:氬氣。6種蘑菇毒素的質譜參數見表1。
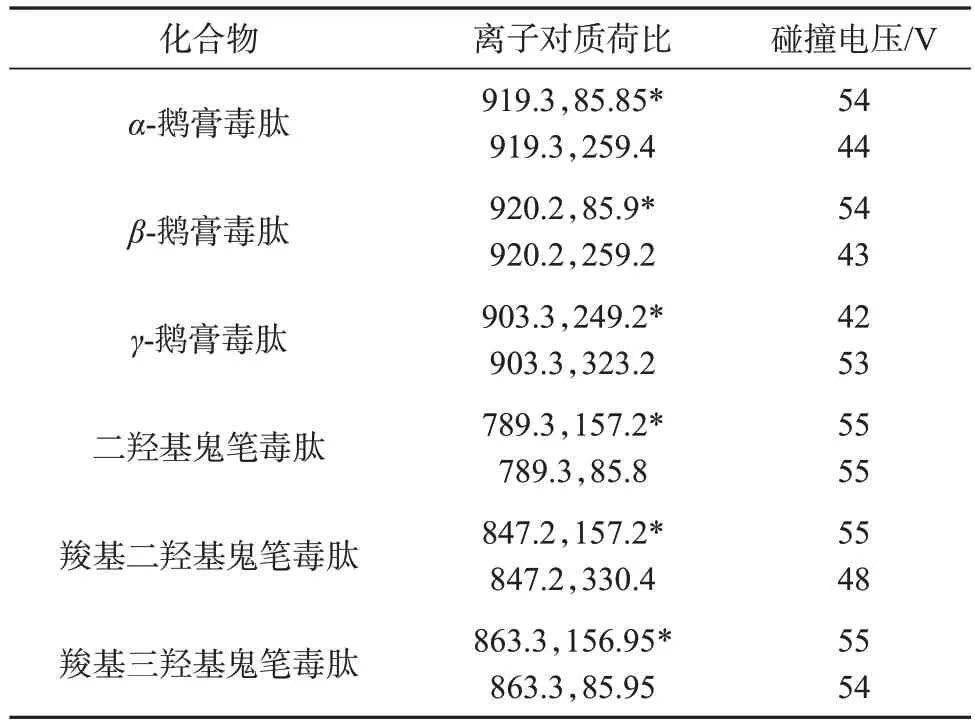
表1 6種蘑菇毒素的質譜參數Tab.1 Mass spectrometry parameters of six mushroom toxins
1.3 標準溶液配制
標準儲備液:分別準確稱取α-鵝膏毒肽、β-鵝膏毒肽、γ-鵝膏毒肽、二羥基鬼筆毒肽、羧基二羥基鬼筆毒肽標準品0.1 mg 于5 只10 mL 容量瓶中,用甲醇定容至標線,配制成質量濃度均為10 μg/mL的標準儲備液。準確吸取羧基三羥基鬼筆毒肽標準溶液0.5 mL 于10 mL 容量瓶中,用甲醇定容至標線,配制成2.5 μg/mL的標準儲備液,各儲備液于-18 ℃冰箱保存。
混合標準中間液:分別準確吸取羧基三羥基鬼筆毒肽標準儲備液4 mL和其它5種標準儲備液各1 mL 于同一只10 mL 容量瓶中,用甲醇定容至標線,配制成各標準品質量濃度均為1 μg/mL的混合標準中間液。
系列混合標準工作溶液:分別準確移取混合標準中間液50、100、200 μL,分別加入950、900、800 μL 體積分數為50%的甲醇水溶液,配制成各化合物質量濃度均分別為50、100、200 ng/mL 的混合標準工作溶液;準確移取質量濃度為100 ng/mL 的混合標準工作溶液50、100 μL,分別加入950、900 μL 50%甲醇水溶液,配制成各化合物質量濃度均分別為5、10 ng/mL的混合標準工作溶液。
1.4 樣品處理
1.4.1 樣品制備
選取適量的干香菇樣品,經粉碎機粉碎,充分混勻后置于潔凈容器中密封并標記,于室溫下避光保存。
1.4.2 樣品提取
準確稱取0.4 g (精確至0.001 g)粉碎后的干香菇樣品,置于15 mL具塞塑料離心管中,加入6.0 mL甲醇,混勻,于渦旋振蕩器上渦旋1 min,超聲提取20 min,冷卻至室溫,以7 000 r/min轉速離心6 min,準確吸取上清液4.0 mL,置于另一離心管中,于45 ℃下氮吹至近干,用4.0 mL水溶解殘渣,加入1.0 mL 正己烷,渦旋1 min,以7 000 r/min 轉速離心6 min,棄去上層正己烷,準確移取水相待凈化液3.0 mL (相當于0.2 g 干香菇樣品)至HLB 固相萃取柱中,以約1 mL/min 的流量全部通過小柱后,用3 mL質量分數為5%甲醇水溶液淋洗、抽干,加入2 mL乙腈-甲醇混合液(體積比為3∶7)洗脫,洗脫液于45 ℃下氮吹至干,準確加入1.00 mL 初始比例流動相,渦旋1 min 溶解殘渣,以濾膜過濾,作為樣品溶液。
2 測量模型
樣品溶液中各組分的質量分數按式(1)計算:
式中:w——蘑菇樣品中目標物質量分數,μg/kg;
ρ——利用標準曲線得到的目標物質量濃度,ng/mL;
V1——提取液體積,mL (V1=6.0 mL);
V2——樣品提取液過凈化柱的體積,mL (V2=3.0 mL);
V3——樣品溶液上機前的定容體積,mL (V3=1.0 mL);
m——蘑菇樣品質量,g。
3 不確定度來源
根據分析方法和實驗步驟,6 個目標物測量結果的不確定度主要來源包括: (1) 測量重復性引入的不確定度; (2) 標準溶液配制過程引入的不確定度; (3) 液相色譜-串聯質譜儀的測量不確定度; (4)樣品處理引入的不確定度; (5) 標準曲線擬合引入的不確定度。對于一些通過實驗證明比較穩定的因素,如樣品的均勻性、實驗室溫度(與校準溫度相差低于2 ℃)等,評定測量不確定度時不予考慮。
4 不確定度評定
因為6 個目標物分析方法相同,測量不確定度評定方法也相同,故以α-鵝膏毒肽為例介紹評定過程。
4.1 測量重復性引入的不確定度
對同一樣品進行6 次獨立重復測量,α-鵝膏毒肽質量分數測定值分別為135.7、134.9、141.2、133.2、139.8、133.3 μg/kg,平均值為136.4 μg/kg,標準偏差s=3.38,重復性測量引入的標準不確定度:
相對標準不確定度:
4.2 標準溶液配制過程引入的不確定度
4.2.1 標準物質純度引入的不確定度
從標準物質證書查閱到α-鵝膏毒肽純度(質量分數)為97.3%,相對擴展不確定度為2% (k=2),則標準物質純度引入的相對標準不確定度:
4.2.4 系列混合標準工作溶液配制過程中引入的不確定度
以200 μL移液器為例,根據JJG 646—2006 《移液器檢定規程》和移液器檢定證書,200 μL 移液器在移取50、100 μL 時,測量結果的相對擴展不確定度為0.7%,k=2,移取200 μL 時,測量結果的相對擴展不確定度為0.4%,k=2,移液器移取不同體積溶液的相對標準不確定度見表2。

表2 系列混合標準工作溶液配制引入的相對標準不確定度分量Tab.2 Relative standard uncertainty component introduced in the preparation of a series of mixed standard working solutions
以1 μg/mL的混合標準中間液配制成不同質量濃度的混合標準工作溶液,采用200 μL和1 000 μL移液器完成稀釋,50、100、200 ng/mL 的混合標準工作溶液稀釋了1 次,5、10 ng/mL 的混合標準工作溶液稀釋了2 次,計算時應重復計算不確定度。根據JJG 646—2006《移液器檢定規程》和移液器檢定證書,200 μL 移液器和1 000 μL 移液器在移取50、100、200 μL時,允許誤差見表2,系列混合標準工作溶液配制引入的相對標準不確定度分量見表2。利用表2數據合成得系列混合標準工作溶液配制過程中引入的相對標準不確定度:
4.2.2 標準物質稱量以及標準儲備液定容體積引入的不確定度
當稱樣量小于5 g 時,天平允許的最大誤差為±0.05 mg,稱取α-鵝膏毒肽標準品1 mg;標準儲備液定容用10 mL容量瓶(A級),參閱文獻[15]可知,A級10 mL 容量瓶的允許誤差為±0.02 mL,由標準物質稱量和標準儲備液定容體積引入的相對標準不確定度:
4.2.3 配制混合標準中間液引入的不確定度
配制混合標準中間液時,使用1 mL A級單標吸量管準確移取1 mLα-鵝膏毒肽標準儲備液于10 mL容量瓶中,用甲醇定容至標線,得到質量濃度為1 μg/mL 的混合標準中間液。參照JJG 196—2006《常用玻璃量器》,1 mL A級單標吸量管、10 mL A級容量瓶的最大允許誤差分別為0.007、0.020 mL,按矩形分布,k= 3,配制混合標準中間液引入的相對標準不確定度:
合成標準物質純度、配制標準儲備液、混合標準中間液、系列混合標準工作溶液引入的各相對標準不確定度分量,得標準溶液配制過程引入的相對標準不確定度:
4.3 液相色譜-串聯質譜儀的測量不確定度
考慮到儀器檢測過程中引入的不確定度,直接引用儀器的檢定證書作為評價依據。儀器的相對擴展不確定度為3.3%,k=2。則儀器檢測過程引入的相對標準不確定度:
4.4 樣品處理引入的不確定度
4.4.1 樣品稱量引入的相對標準不確定度
天平校準允許的最大誤差為±0.000 05 g,樣品稱取質量(m)為0.4 g,按矩形分布,k= 3,樣品稱量引入的相對標準不確定度:
4.4.2 樣品提取引入的相對標準不確定度
分析樣品處理步驟,用10 mL B 級流出式分度吸量管吸取6.0 mL甲醇,5 mL B級流出式分度吸量管吸取4.0 mL上清液,5 mL B級流出式分度吸量管吸取4.0 mL 水,5 mL B 級流出式分度吸量管吸取3.0 mL質量分數為5%甲醇水溶液,1 000 μL移液器吸取1.00 mL 初始比例流動相,根據JJG 196—2006《常用玻璃量器檢定規程》可以計算由樣品提取引入的相對標準不確定度:
由于上述各因素互不相關,樣品處理過程引入的不確定度:
4.5 標準曲線擬合引入的相對標準不確定度
以目標物質量濃度為橫坐標,目標物的色譜峰面積比為縱坐標,按照最小二乘法進行線性擬合,得標準工作曲線線性方程為y=82.156 5x-322.693。標準工作曲線擬合引入的標準不確定度u(L)按式(2)、式(3)計算[15-16]:
式中:sR——標準工作曲線的標準偏差;
b——標準工作曲線的斜率;
p——樣品平行測定的次數,p=6;
n——標準溶液濃度點,n=5;
x0——樣品溶液質量濃度,ng/mL;
xˉ——系列混合標準溶液的平均質量濃度,73 ng/mL;
yi——第i標準點的色譜峰面積;
xi——第i標準點的質量濃度ng/mL;
a——標準工作曲線的截距。
樣品平行測定6 次,樣品溶液中α-鵝膏毒肽質量濃度分別為27.14、26.97、28.23、26.65、27.96、26.65 ng/mL,x0=27.27 ng/mL。將相關數據代入式(2)、式(3),得sR為0.329 5,標準工作曲線擬合引入的標準不確定度u(L)=0.292 9 ng/mL,則標準工作曲線擬合引入的相對標準不確定度:
4.6 合成標準不確定度評定及擴展不確定度
以上各不確定度分量互不相關,因此蘑菇中α-鵝膏毒肽測定結果的合成相對不確定度:
取置信水平P=95%,包含因子k=2,α-鵝膏毒肽含量測定結果的相對擴展不確定度按照Urel=k×urel計算。β-鵝膏毒肽、γ-鵝膏毒肽、二羥基鬼筆毒肽、羧基二羥基鬼筆毒肽和羧基三羥基鬼筆毒肽依據同樣的不確定度評定過程,評定結果見表3。

表3 6種蘑菇毒素合成標準不確定度和檢測結果報告Tab.3 Combined standard uncertainty and test result report of six mushroom toxin
5 結語
根據BJS 202008《蘑菇中α-鵝膏毒肽等6 種蘑菇毒素的測定》,對蘑菇中6 種蘑菇毒素進行測定,并對測定結果的不確定度進行評定。樣品重復測量和標準溶液配制過程引入的相對標準不確定度最大,其次是標準曲線擬合引入的不確定度,其它影響因素較小。在實際工作中,需要定期對移液器和液相色譜-串聯質譜儀進行校準和維護保養;購買具有合格檢驗證書的標準品和樣品移取用具;同時提升檢測人員的操作規范和穩定性,熟悉檢測方法,提高檢測結果的準確性。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·八年級物理人教版(2019年9期)2019-11-25 07:33:02
當代陜西(2019年8期)2019-05-09 02:22:48
中學生數理化·八年級物理人教版(2019年3期)2019-04-25 06:20:54
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
中學生數理化·八年級物理人教版(2018年3期)2018-05-31 08:52:45
家庭影院技術(2018年4期)2018-05-09 07:07:52
數學小靈通(1-2年級)(2017年10期)2017-11-08 08:39:45
少兒科學周刊·兒童版(2016年1期)2016-03-14 03:52:21
專用汽車(2016年4期)2016-03-01 04:13:43
