高效液相色譜法測定化妝品原料中N-乙酰神經氨酸
2024-02-20 09:12:38刁飛燕李俊婕吳曉云劉慧香于海英李啟艷
化學分析計量 2024年1期
刁飛燕,李俊婕,吳曉云,劉慧香,于海英,李啟艷
(山東省食品藥品檢驗研究院,國家藥品監督管理局化妝品原料質量控制重點實驗室,特色植物資源化妝品濟南市工程研究中心,產業技術基礎公共服務平臺,濟南 250101)
N-乙酰神經氨酸(Neu5Ac),通用名為唾液酸(SA)。唾液酸是一類母體結構為9個碳原子組成的神經氨酸,是廣泛存在于生物系統中的一類天然糖類化合物。在大部分哺乳動物中發現的唾液酸主要是N-乙酰神經氨酸,因此常把N-乙酰神經氨酸稱為唾液酸[1-2],已被證實其具有抗病毒、抗氧化、抗衰老等多種生理功效[3-5],在化妝品中常用作保濕劑、皮膚保護劑等。燕窩、牛奶、乳制品等是N-乙酰神經氨酸的主要天然來源[6-8]。N-乙酰神經氨酸在2021年通過化妝品新原料注冊備案,成為國產化妝品新原料。
目前N-乙酰神經氨酸的測定法主要有紫外可見分光光度法[9-10]、高效液相色譜法[11-12]、液相色譜-串聯質譜法[13-14]。比色法中采用間苯二酚顯色法測定N-乙酰神經氨酸含量,操作繁瑣,化妝品原料基質復雜,測定單一組分時易受干擾,導致測定結果偏高;而液相色譜-質譜法測定成本相對較高。目前尚無針對化妝品原料中N-乙酰神經氨酸測定的標準方法,難以保證原料的有效性、安全性。筆者選擇強陽離子SCX 色譜柱,優化樣品處理條件,利用高效液相色譜-二極管陣列檢測器進行測定,解決了該原料極性大,在一般反相色譜柱中不能保留的難題,為化妝品原料中N-乙酰神經氨酸的質量檢測提供了技術支持。
1 實驗部分
1.1 主要儀器與試劑
高效液相色譜儀:LC-20AT 型,日本島津實驗器材有限公司。
電子天平:MS105DU 型,感量為0.01 mg,瑞士梅特勒托利多公司。
純水儀:Milli-Q A10型,美國密理博公司。
數控超聲波清洗器:KQ-500DE型,昆山市超聲儀器有限公司。
數顯恒溫水浴鍋:HH-S6 型,山東博科科學儀器有限公司。
乙腈、甲醇:色譜純,德國默克公司。
磷酸、乙酸:色譜純,天津市科密歐化學試劑有限公司。
N-乙酰神經氨酸對照品:純度(質量分數)為98%,美國西格瑪奧德里奇公司。
供試品原料:共11批,某企業提供。
實驗用水為超純水,由密理博純水儀制得。
1.2 色譜條件
色譜柱:CAPCELL PAK SCX 柱(250 mm×4.6 mm,5 μm,日本資生堂公司);檢測器:二極管陣列檢測器;流動相:乙腈-0.1%磷酸溶液(體積比為80∶20),流量為1.0 mL/min;檢測波長:205 nm;柱溫:35 ℃;進樣體積:10 μL;
1.3 溶液配制
N-乙酰神經氨酸對照品儲備液:2 000 μg/mL,稱取N-乙酰神經氨酸對照品適量,用2% (體積分數,下同)乙酸溶液稀釋并定容。
N-乙酰神經氨酸系列標準工作溶液:質量濃度分別為10.0、20.0、50.0、100.0、250.0、500.0 μg/mL,分別準確移取不同體積的N-乙酰神經氨酸對照品儲備液,用2%乙酸溶液稀釋制得。
樣品溶液:稱取供試品原料適量(其中含N-乙酰神經氨酸約40 mg),精密稱定,置于25 mL 比色管中,加入50% (體積分數,下同)乙酸溶液10 mL 溶解,蓋上玻璃塞,置于80 ℃水浴中加熱30 min,取出比色管,冷卻至室溫。將水解液轉移至250 mL容量瓶中,用水稀釋至標線,混勻,過孔徑為0.45 μm 濾膜,取續濾液作為樣品溶液。
空白對照溶液:除不加供試品外,其余測定步驟與樣品溶液制備步驟一致。
1.4 實驗方法
取N-乙酰神經氨酸系列標準工作溶液及樣品溶液,按1.2色譜條件進樣分析,記錄色譜峰面積,建立標準工作曲線,按色譜峰面積外標法計算樣品中的N-乙酰神經氨酸含量。
2 結果與討論
2.1 色譜條件選擇
2.1.1 色譜柱
分別考察Symmetry Shield RP18 柱(簡稱C18柱)、CAPCELL PAK SCX 柱( 簡 稱SCX 柱) 和Hypersil GOLD AQ柱(簡稱AQ柱)對N-乙酰神經氨酸的保留行為。結果表明,C18柱和AQ柱對N-乙酰神經氨酸不易保留;SCX柱使用混合強陽離子交換填料,具有離子交換和疏水作用雙重功能,選擇性強,延長了N-乙酰神經氨酸的保留時間,能實現對N-乙酰神經氨酸的良好分離,且色譜峰形良好,故選擇CAPCELL PAK SCX 柱(250 mm×4.6 mm,5 μm)作為分析柱。
2.1.2 流動相
分別考察甲醇-0.1% (體積分數,下同)磷酸溶液、乙腈-0.1%磷酸溶液、乙腈-0.1% (體積分數,下同)甲酸溶液3種流動相體系。結果表明,與甲醇相比,有機相選擇乙腈,色譜圖中雜質峰較少;水相選擇水或0.1%甲酸溶液時,峰形較寬并前延,水相為0.1%磷酸溶液時,色譜峰形得到改善。
考察乙腈與0.1%磷酸溶液的體積比分別為90∶10、80∶20、70∶30對測定結果的影響。結果顯示,隨著水相比例的增加,N-乙酰神經氨酸的保留時間延長。當乙腈與0.1%磷酸溶液的體積比為80∶20時,保留時間及峰形佳,最終選擇流動相組成為乙腈-0.1%磷酸溶液(體積比為80∶20)。
2.1.3 檢測波長
采用高效液相色譜儀對N-乙酰神經氨酸標準工作溶液在190~400 nm 波長范圍內進行掃描,所得光譜圖為典型的末端吸收曲線,參考最大吸收檢測波長,選擇檢測波長為205 nm。
2.2 樣品處理方法
唾液酸可分為游離態唾液酸和結合態唾液酸兩種形式,其中游離態唾液酸是未與糖或蛋白結合的唾液酸,樣品中結合態的唾液酸經過加熱及酸處理,產生游離態的唾液酸[15],超聲提取不能水解完全,需將樣品水浴加熱后測定,因此采取水解樣品處理方法,并考察不同水解溶劑、水解溫度、水解時間三個因素對提取結果的影響。
分別選擇50%乙酸溶液、0.5 mol/L硫酸氫鈉溶液、0.1 mol/L 硫酸溶液、1% (體積分數)磷酸溶液4種水解液進行試驗。結果表明,在相同溫度、相同水解時間條件下,以50%乙酸溶液和1%磷酸溶液為水解溶劑時,N-乙酰神經氨酸含量測定值高于其余兩種水解溶劑,但經1%磷酸溶液水解后樣品溶液顏色明顯加深,雜質的色譜響應值增大,目標物檢出限略高,故選擇50%乙酸溶液為水解溶劑。
分別選擇水解溫度為30、40、60、80、100 ℃進行試驗,結果如圖1所示。由圖1可以看出,隨著水解溫度升高,目標物測定值逐漸增大,當水解溫度超過80 ℃時,目標物發生部分降解,測定值逐漸降低,故選擇水解溫度為80 ℃。

圖1 不同水解溫度時的測定結果Fig.1 Determination results of diffferent hydrolysis temperature
分別選擇水解時間為10、20、30、40、60 min 進行試驗,結果如圖2所示。由圖2可以看出,當水解時間為10~30 min 時,目標物測定值逐漸增大;當水解時間為30~60 min 時,目標物測定值基本一致,故選擇水解時間為30 min。

圖2 不同水解時間時的測定結果Fig.2 Determination results of different hydrolysis time
2.3 方法專屬性
分別取空白對照溶液、N-乙酰神經氨酸標準工作溶液和樣品溶液,按1.2色譜條件進行測定,記錄色譜圖,如圖3所示。

圖3 空白對照溶液、 N-乙酰神經氨酸標準溶液和樣品溶液色譜圖Fig.3 Chromatograms of blank control solution,N-acetylneuraminic acid standard solution and sample solution
由圖3 可以看出,N-乙酰神經氨酸保留時間約為6.8 min,色譜峰形良好。空白對照溶液及樣品溶液基質均無干擾,滿足試驗要求,表明該方法專屬性良好。
2.4 線性方程與檢出限、定量限
取N-乙酰神經氨酸系列標準工作溶液,按照質量濃度由低到高的順序依次進樣分析,以溶液質量濃度為橫坐標(x),以色譜峰面積為縱坐標(y),進行線性回歸,計算線性方程和相關系數。結果顯示N-乙酰神經氨酸質量濃度在10.0~500.0 μg/mL 范圍內與色譜峰面積線性關系良好,線性方程為y=5 420.15x-1 181.45,相關系數為0.999 9。
取N-乙酰神經氨酸質量濃度為10 μg/mL 的標準工作溶液,用2%乙酸溶液逐級稀釋,分別以3 倍信噪比和10 倍信噪比對應的質量濃度作為檢出限和定量限,該方法檢出限為0.12 μg/mL,定量限為0.4 μg/mL。
2.5 加標回收與精密度試驗
稱取樣品適量(其中含N-乙酰神經氨酸約20 mg),精密稱定,分別進行低、中、高三個濃度水平的加標回收及精密度試驗,每個濃度水平平行制備6份,按1.3方法制備加標樣品溶液,按1.2色譜條件分別測定,計算N-乙酰神經氨酸的回收率及相對標準偏差,結果見表1。由表1 可知,目標組分的平均回收率為97.7%~98.9%,測定結果的相對標準偏差(RSD)為0.37%~1.36%,表明該方法準確度及精密度良好,滿足分析要求。

表1 加標回收與精密度試驗結果Tab.1 Results of recovery and precision test
2.6 穩定性試驗
按1.3 方法平行制備樣品溶液,分別于第0、2、4、8、12、24 h 進樣測定,計算色譜峰面積,結果見表2。由表2可知,N-乙酰神經氨酸色譜峰面積測定結果的相對標準偏差為0.90%,表明樣品溶液在24 h內穩定性良好。

表2 穩定性試驗結果Tab.2 Results of stability test
2.7 樣品測定
目前國家藥品監督管理局公布的化妝品新原料備案信息中顯示,N-乙酰神經氨酸在化妝品中使用時的最大允許質量分數為2%,故檢測原料中N-乙酰神經氨酸的含量對保障化妝品的質量及安全使用具有重要意義。N-乙酰神經氨酸制備方法主要是天然產物提取、化學合成法、酶法合成[16]、微生物發酵法[17]。備案新原料N-乙酰神經氨酸的制備方法為微生物發酵法,含量可達98%以上。《已使用化妝品原料目錄(2021 年版)》收錄的燕窩提取物中含有N-乙酰神經氨酸,因此選取燕窩提取物和N-乙酰神經氨酸兩類原料進行測定。
按所建方法測定11 批供試品原料中N-乙酰神經氨酸的含量,其中1#~8#原料標示為燕窩提取物,9#~11#標示為N-乙酰神經氨酸,測定結果見表3。由表3 可知,所測樣品中N-乙酰神經氨酸的質量分數范圍為6.2%~98.5%。6#、8#原料中N-乙酰神經氨酸的含量測定值略低于標示值,其余樣品測定值與標示值相符。
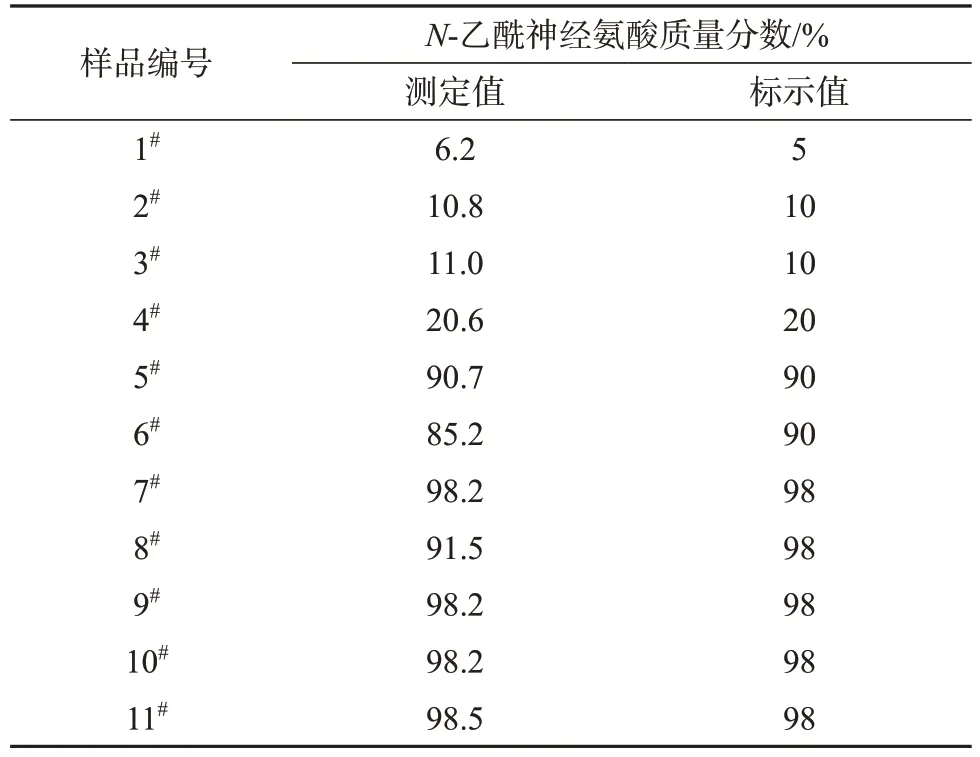
表3 樣品測定結果Tab.3 Determination results of sample
3 結語
建立了檢測化妝品原料中N-乙酰神經氨酸的高效液相色譜方法。該法準確、高效,未使用有機溶劑衍生,大幅降低了對人員和環境的影響。該法具有良好的線性、準確度、精密度,為制定化妝品原料中N-乙酰神經氨酸的含量測定相關標準提供了依據。
