柴油車尾氣氧化催化劑硫磷中毒研究進展
2024-02-20 13:10:16劉叢瑋單文坡
石油煉制與化工 2024年2期
關鍵詞:催化劑
劉叢瑋,王 猛,張 燕,3,單文坡,3
(1.中國科學院城市環境研究所,中國科學院區域大氣環境研究卓越創新中心,福建 廈門 361021;2.中國科學院大學;3.中國科學院城市環境研究所寧波觀測研究站,浙江省城市環境過程與污染控制重點實驗室)
《2022年交通運輸行業發展統計公報》顯示,以柴油機為主要動力來源的公路運輸占我國貨運總量的72.3%,且在今后相當長時間內仍然無法被完全取代[1]。柴油車尾氣中含有大量污染物,《中國移動源環境管理年報(2022)》統計顯示,2021年柴油車的一氧化碳(CO)、碳氫化合物(HC)、氮氧化物(NOx)和顆粒物(PM)的排放量分別為1 187,183,5 021,64 kt,是我國大氣污染的主要來源,會導致灰霾、光化學煙霧等多種嚴重的環境危害[2]。因此,有效控制柴油車污染物排放具有重要意義。
我國柴油車尾氣排放標準日益嚴格,單一的后處理技術已無法達到現行排放標準的要求,需要耦合多種后處理技術對尾氣進行處理。滿足國Ⅵ排放標準的柴油車后處理技術路線包括柴油車尾氣氧化催化劑(DOC)、柴油顆粒捕集器(DPF)、氨選擇性催化還原(NH3-SCR)技術和氨氧化催化劑(AOC)(如圖1所示)[3]。DOC的主要作用是催化凈化CO和HC,并氧化部分一氧化氮(NO)為二氧化氮(NO2)[4]。DPF利用相鄰捕集器孔道前后交替封堵,使尾氣從壁面穿過,從而實現尾氣中PM的截留捕集。NH3-SCR技術是利用還原劑氨氣(NH3)選擇性地將NOx催化還原為氮氣(N2)[5]。AOC主要用于去除為保障NOx凈化效率而過量噴射的NH3[6]。
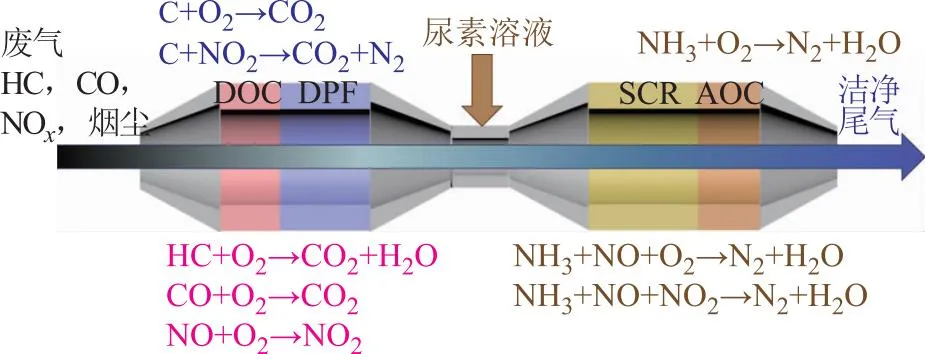
圖1 國Ⅵ柴油車后處理系統[3]
位于整個后處理系統最前端的DOC主要包括:殼體、減振層、蜂窩載體和催化劑涂層,其中以貴金屬Pt、Pd為活性組分的催化劑涂層是核心[7]。DOC的作用至關重要,除了催化氧化尾氣中CO和HC使其滿足排放標準,還能為下游PM消除和NOx消除提供足量的NO2。一方面NO2可降低碳煙燃燒溫度,有效提高DPF被動再生效率;另一方面當NO2/NO體積比為1時,SCR催化劑可實現快速SCR反應,極大提高低溫下的脫硝效率。可見,DOC是有效控制柴油車污染物排放的關鍵。
柴油和潤滑油中存在一定量的含硫、含磷有機物,這導致柴油車尾氣中不可避免地含有硫、磷等組分。硫、磷極易毒化催化劑,且上游的DOC首當其沖,繼而造成整個后處理系統的凈化效率降低。關于DOC的催化劑材料、反應機理、硫中毒問題等已有多篇文獻綜述,但很少有學者系統地關注硫磷中毒的影響及緩解策略[4,7-11]。本文從硫磷中毒狀況、中毒機理和緩解中毒策略等方面系統綜述DOC催化劑硫、磷及硫磷復合中毒的研究現狀,總結DOC催化劑當前面臨的挑戰,并在此基礎上提出未來需要重點關注的研究方向。
1 硫中毒
1.1 硫的來源及硫中毒失活
柴油組分中存在一定量的含硫有機物(主要為噻吩類),這類物質在燃燒過程中會產生硫化氫(H2S)和二氧化硫(SO2),柴油車尾氣在富氧條件下燃燒的主要產物為SO2[12]。盡管已經限制了柴油中的硫含量,比如國Ⅵ標準中要求柴油中硫質量分數需控制在10 μg/g以下,但是由于政策落實差異、技術迭代周期等問題,柴油車尾氣中仍然有一定量的SO2存在[13-14]。即使使用低硫柴油,仍不能完全規避SO2的生成,尾氣中依舊含有體積分數為0.000 5%~0.001%的SO2[9]。
表1列舉了DOC催化劑硫中毒前后的活性變化情況。顯而易見,無論是粉末催化劑還是整體催化劑,硫的存在均會對其氧化CO、丙烯(C3H6)、NO的性能產生顯著影響。硫中毒的程度與催化劑貴金屬類型、含量及載體等因素息息相關。與Pt基催化劑相比,Pd基催化劑硫中毒現象更為顯著。Kim等發現,硫化后Pt/Al2O3和Pd/Al2O3催化劑CO轉化50%的溫度(T50)均升高,但后者的升高幅度(90 ℃)遠高于前者(50 ℃)[15]。在商用整體催化劑上,硫沉積在催化劑表面,繼而毒化催化劑。Kr?cher等研究表明,DOC上的硫沉積質量可以高達涂層總質量的4.7%,NO的最大轉化率下降15%[16-17]。

表1 催化劑硫中毒前后活性對比
1.2 硫中毒對催化劑的影響
1.2.1硫對活性組分的影響
DOC催化劑的主要活性組分為貴金屬Pt、Pd,SO2在DOC催化劑表面發生如下反應[27-28]:

(1)
(2)
(3)
(4)
(5)
*表示貴金屬位點。
CO、C3H6與O2在催化劑表面的反應過程如下[29]:
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
由上述反應方程可知,SO2在表面氧化過程中會和CO及C3H6產生強烈的競爭作用,主要是對貴金屬位點以及活性氧的競爭,從而抑制催化劑對CO,C3H6,NO的氧化[9,11]。更為甚之,SO2可與貴金屬直接形成硫酸鹽物種,或者以硫酸鹽形式沉積在DOC表面,從而間接影響貴金屬性質,進而導致貴金屬硫中毒,使得DOC失活。試驗和密度泛函理論(DFT)計算結果均表明,在富氧和SO2存在的狀態下,Pd傾向于形成低活性的硫酸鈀物種,該物種將會導致催化劑中毒失活,而Pt傾向于以金屬態存在,不易形成穩定的硫酸鹽(如圖2所示),因此,Pd較Pt更容易產生硫中毒現象[26,30-31]。雖然Pt不易形成硫酸鹽,但硫的存在會降低貴金屬的表面能,進而誘導Pt顆粒生長,使其更容易團聚[32-34]。Andersson等研究發現,在250 ℃硫化25 h后,Pt團聚現象嚴重,顆粒由2.1~3.2 nm劇增至100 nm以上[35]。此外,硫在催化劑表面會形成新的Br?nsted酸位點,Pt、Pd貴金屬的電荷向酸性位點偏移,導致表面處于相對缺電子的狀態,對CO和O2的吸附能力大幅降低,對C3H6的吸附增強,產生自抑制效應,致使催化劑氧化CO和C3H6性能大幅降低[22,36-37]。

圖2 Pd、Pt催化劑硫中毒機理示意[30]
1.2.2硫對載體的影響
尾氣中的SO2會和載體結合形成硫酸鹽或者亞硫酸鹽,堵塞孔道,降低載體的比表面積,影響污染物在催化劑上的傳質過程[23-24,38-39]。載體硫化的途徑主要有兩種:一是氣體中的SO2直接和載體反應形成亞硫酸鹽;二是SO2被貴金屬氧化后形成三氧化硫(SO3)或硫酸,上述物質與表面金屬形成硫酸鹽。前者形成的亞硫酸鹽不穩定,在低溫下易分解,而后者形成的硫酸鹽更多、更穩定,對載體物理性質的影響更顯著[34]。載體上硫酸鹽堆積量過大時,會從載體反向溢出至貴金屬表面,進而直接遮蔽催化劑的活性位點。載體上硫酸鹽的形成及堆積與貴金屬類型、比例、粒徑等因素密切相關。常用的貴金屬Pt、Pd中,Pd會加劇載體的中毒狀況,這是由于Pd對亞硫酸鹽的氧化能力更強,在相同溫度下會形成較Pt催化劑更多的硫酸鹽物種,加劇硫在催化劑表面的沉積。Wilburn等研究發現,相同毒化條件下,高Pd含量(Pd與Pt摩爾比為0.7∶0.3)催化劑上堆積的硫酸鹽含量是低Pd含量(Pd與Pt摩爾比為0.15∶0.85)催化劑的2.5倍以上[40]。此外,研究表明,在相同毒化條件下,Pt粒徑為3.9 nm的Pt/Al2O3催化劑表面硫酸鹽含量較Pt粒徑為10.4 nm時增加了2倍,這是因為貴金屬的粒徑越小,越易促使載體上的亞硫酸鹽向硫酸鹽的轉化[41]。
1.2.3硫對整體催化劑的影響
在尾氣中僅存在硫的情況下,硫一般均勻分布于催化器件上,這是由于硫中毒具有一定的可逆性,在硫沉積與分解的過程中,造成了涂層的緩慢硫化[16]。實際工況下,硫的分布會受到尾氣中磷含量、催化劑涂層配方等因素的影響,該內容將在硫磷復合中毒章節進行詳細綜述。
1.3 緩解硫中毒策略
緩解硫中毒的策略主要包括提升催化劑抗硫性能和硫中毒后再生性能兩方面,前者通過對催化劑進行改性以提升其抗硫性能,后者通過采用加熱手段分解催化劑表面堆積的硫酸鹽,使催化劑活性得到恢復。
1.3.1抗硫催化劑設計
抗硫DOC的設計主要采用貴金屬聯用、載體改性、添加助劑等方式,削弱硫和DOC之間的相互作用,提升其抗硫性能。將Pt、Pd貴金屬聯用,SO2會優先吸附在Pt位點上,保護Pd位點不被毒害,吸附在Pt位點上的SO2會以分子態而非亞硫酸鹽、硫酸鹽的形式存在,更容易脫附。同時,Pd和Pt形成的Pt-Pd合金可以進一步削弱分子態SO2和貴金屬的相互作用,從而使其在吸附后很容易從表面脫除[42-44]。
酸性載體,如ZrO2、SiO2,可以有效避免硫的堆積。Shen Meiqing等研究發現,使用Ce-Zr載體可以減少載體上硫的堆積,且堆積的硫主要以亞硫酸鹽的形式存在,更容易分解,抗硫性有一定程度的提高[45]。Kim等利用溶膠凝膠法將SiO2和ZrO2混合,使ZrO2保持一定酸性的同時,降低了其表面的堿度,提高了催化劑的抗硫性[46]。將TiO2作為載體,硫可以與其形成易分解的硫酸鈦,減輕硫在催化劑表面的中毒效應[10]。由于TiO2本身已經擁有較為優異的抗硫性,因此對以TiO2為載體的催化劑的研究集中在保留抗硫性的同時,提高其活性。Zhang Na等在TiO2中摻雜Ce,增強了催化劑的儲/放氧能力,在擁有較好抗硫性的同時,還提高了催化劑的催化性能[47]。Yang Zhengzheng等發現,Y作為結構助劑摻雜進TiO2可以維持其銳鈦礦結構,并提高Pt的分散度,使其具有較強抗硫能力的同時,進一步提升了催化活性[48]。Zhou Jiali等將Pt粒子封裝到TiOx載體內,在保留TiO2本身優異抗硫性的基礎上,使Pt穩定在具有高催化活性的Pt0狀態,提高了催化活性[49]。
添加助劑也是一種常用的抗硫方法,如直接將硫自身作為一種助劑添加至催化劑組分中形成新的Br?nsted酸位點,可以提升催化劑的酸性,減少硫在催化劑表面的沉積[50]。過渡金屬V和W常作為酸性助劑,用以增強催化劑的酸性,提升催化劑的抗硫性能[20,51]。一些非酸性過渡金屬可以通過削弱硫和催化劑的相互作用以實現抗硫性能的提升,如Fe作為助劑可以削弱SO2和載體之間的相互作用,有效抑制Al2O3向Al2(SO4)3的轉變,從而實現抗硫性能的提升[15]。Xia Meirong等用理論計算的方式證明,將Mo和Pt結合形成合金,可降低SO2在貴金屬上的吸附能力,提高催化劑抗硫中毒能力[52]。Franken等將Mn和Pt形成合金,用來削弱硫和貴金屬之間的相互作用,實現催化劑抗硫性能的提升[21]。
1.3.2硫中毒再生

還原性氣體的種類、濃度以及還原溫度均可影響再生效果。Luo Jinyong等研究發現,氫氣(H2)具有較強的還原能力,它的脫硫效果較其他還原劑(如CO)更好,在500 ℃下使用CO還原可以去除40%的SO2,而使用H2再生可以去除80%的SO2[39]。Galisteo等研究發現,在600 ℃下用H2還原Pt/Al2O3,硫主要以SO2的形式脫去,催化劑活性可以緩慢恢復[36]。但過量的H2會深度還原已分解的SO2,繼而生成惡臭物質H2S,因此再生過程中需要合理控制還原劑的濃度。雖然還原性氣氛可以有效提升催化劑的再生效果,但在實際應用中難以在車上外加還原劑。因此,研究者們也嘗試了一些易于實現的再生手段。Li Yuan等發現柴油車尾氣中的NO可有效遏制貴金屬位點周圍的硫氧化物(SOx)轉化為穩定吸附的硫酸鹽,還可促使已吸附的硫酸鹽向非穩態的中間產物轉化并脫附,從而實現催化劑的再生[38]。Millo等發現通過貧燃和富燃氣氛交替吹掃,催化劑表面的硫幾乎可以完全脫附,這表明通過精確控制發動機可對硫中毒催化劑進行有效熱再生[54]。
盡管對DOC的硫中毒機理及其緩解策略等研究已較為充分,但是在實際應用過程中,DOC硫中毒仍然是影響其使用壽命的重要因素之一,這可能歸因于以下幾方面:①催化劑的抗硫性有所改進,但仍無法避免硫酸鹽長時間的累積;②部分抗硫改性未綜合考察催化劑其他方面的性能,如耐熱性;③再生溫度高或需外加還原劑等原因導致催化劑在實際應用過程中難以有效再生。因此,后續仍需開發高效耐久且易于應用的抗硫催化材料,并且DOC再生技術需要向原位再生技術轉型。
2 磷中毒
2.1 磷的來源及磷中毒失活
磷的主要來源是燃料雜質和潤滑油添加劑(如二烷基二硫代磷酸鋅),這些組分的含量在前端控制過程中沒有明確的規范和技術標準去約束[14]。在尾氣排放過程中,含磷物質或是以磷酸鹽的形式附著在顆粒以及液滴上,或是直接以氣態磷酸的形式隨尾氣排出[17]。表2列出了部分文獻中報道的整體式及粉末催化劑磷中毒后的失活狀況。磷中毒后,粉末催化劑和整體催化劑活性均有一定的下降,特別是在部分Pt、Pd分區涂覆的整體式催化劑中,磷會沉積在Pt含量較高的進氣端,嚴重降低催化劑的NO氧化活性,Agote-Arán等取樣的分區涂覆DOC中,磷中毒后NO的最大轉化率從63%下降至29%[55-57]。
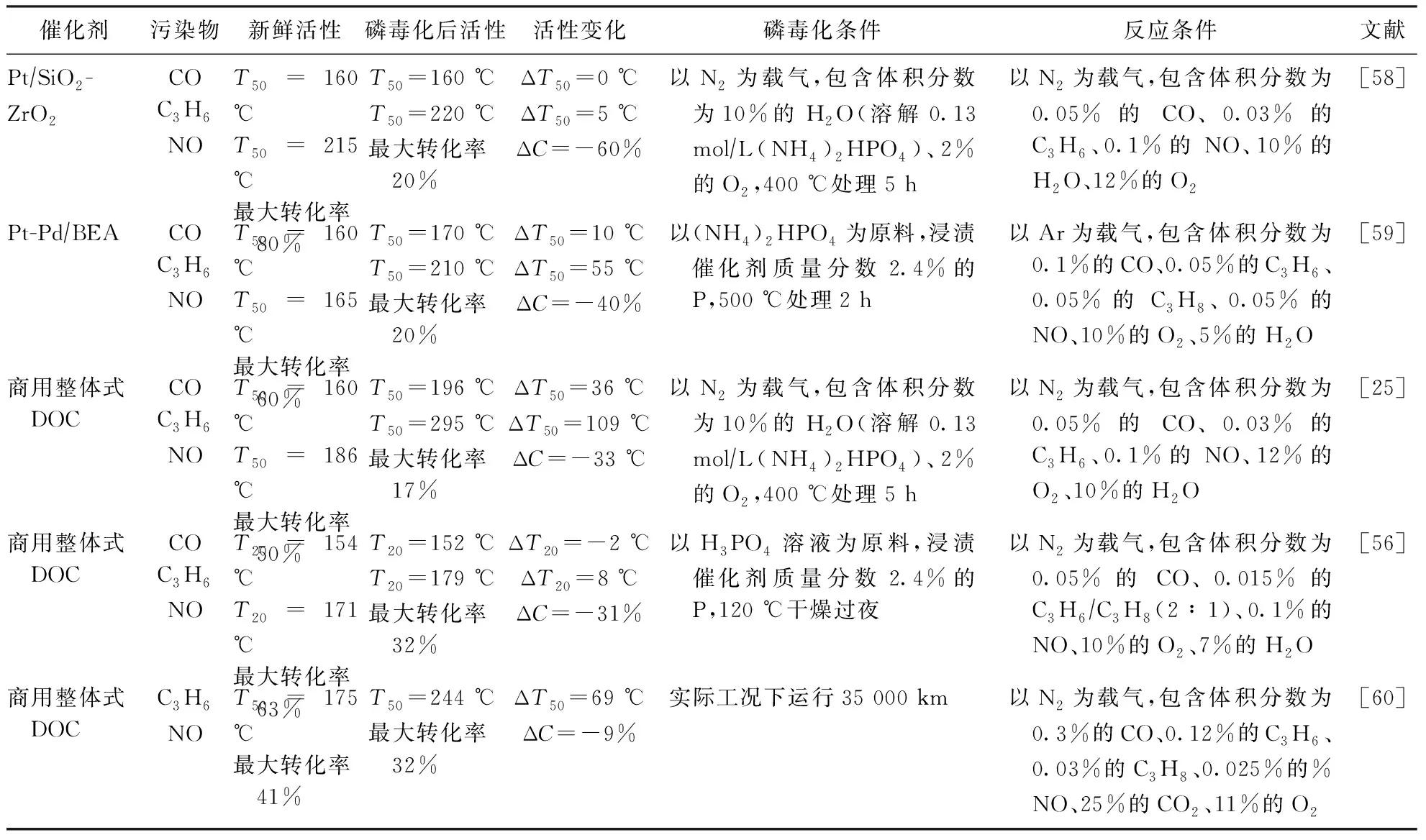
表2 催化劑磷中毒前后活性對比
2.2 磷中毒對催化劑的影響
2.2.1磷中毒對活性組分的影響
磷會改變貴金屬顆粒的形貌。Matam等發現,磷沉積在配位不飽和的Pt粒子上,并與其邊緣產生相互作用,使Pt顆粒由原來的立方顆粒變為球形顆粒[61]。Honkanen等在整體式催化劑上也發現類似現象,經過磷處理的催化劑,貴金屬由原來的不規則形狀變成了球形[62]。而NO的氧化是一種結構敏感反應,球形催化劑活性表面積較小,且對NO的氧化活性遠低于不規則形催化劑,導致DOC的催化活性下降[61]。磷會誘導貴金屬氧化,使擁有更高催化活性的低價態貴金屬向低活性的高價態Pd2+、Pt4+磷酸鹽轉化,這是DOC催化劑磷中毒失活的另一個重要原因[63-65]。Bergman等進一步研究發現,在Pd和Pt中,磷會優先和Pt結合,改變貴金屬的電子結構和周圍的電荷分布,使Pt傾向于以氧化態形式存在。根據圖3可以看到,在沒有Pt分布的區域,磷分布較少且較平均,但是在有Pt分布的區域,磷的分布和Pt的分布有明顯的相關性,磷對貴金屬Pt有更明顯的毒化作用[66]。

圖3 STEM 和EDS探測得到的Al,O,P,Pt,Pd分布[66]
2.2.2磷中毒對載體的影響
2.2.3磷對整體式催化劑的影響
對整體式DOC而言,磷中毒主要分布在組件的前端,Agote-Arán等對磷中毒的DOC從進氣口到出氣口分6段進行取樣,發現磷中毒在進氣口最為嚴重[63]。在實際運行的老化的整體式催化劑中,往往會同時存在硫磷污染物,二者的分布會相互影響,此內容將在硫磷復合中毒章節進行具體論述。
2.3 緩解磷中毒策略
DOC磷中毒再生及抗磷性研究較少,但是在汽油車尾氣處理催化劑或天然氣汽車尾氣處理催化劑中,催化劑載體多以鋁為主,活性組分主要以貴金屬Pt、Pd為主[72-73],其相關研究可供參考。
2.3.1抗磷改性
目前,關于DOC抗磷改性的研究較少,但在催化劑組成較接近的天然氣汽車三效催化劑的研究中,Zhang Guochen等利用磷酸鈰、磷酸釔對載體改性,發現磷酸鈰摻雜的Pd/Al2O3磷中毒后CH4的T50僅提升5 ℃,而未改性的催化劑T50提升了30 ℃,催化劑的抗磷性能顯著提升。這是由于磷酸鈰抑制了Al2O3載體在磷中毒過程中的相變,避免其形成低比表面積、低活性的多層方晶石型磷酸鋁,同時抑制了磷在催化劑表面的堆積,減少因磷中毒導致的Pd分散度的降低[74-75]。在對DOC的相關研究中,Agote-Arán提出了與天然氣汽車抗磷中毒方式接近的觀點,對磷中毒較為集中的DOC進氣端進行微磷化以提高抗磷中毒性能,但結果還有待進一步驗證[55]。
2.3.2磷中毒再生
DOC上的磷難以通過熱分解的方式再生。Andersson等嘗試在700 ℃的還原氣氛下再生已行駛80 000 km和160 000 km的DOC,發現其表面沉積的磷難以去除[35]。目前,關于DOC磷中毒再生的研究較少,主要再生方法可借鑒組分類似的汽油車三效催化劑體系,最常用的再生方式為清洗。清洗劑主要以弱酸清洗劑為主,這類清洗劑往往可以同時去除多種中毒物質(硫、磷、鈣、鎂等),但是可能會溶解一定的涂層組分,因此需要控制酸的用量,并且酸洗也無法恢復已經燒結聚集的活性組分,因此后續大多使用有機弱酸,避免清洗劑對涂層的損害。同時,部分有機弱酸可以輔助熱再生過程中貴金屬的再分散[76]。Cabello等用檸檬酸溶液對DOC進行清洗,發現檸檬酸溶液可以破壞催化劑表面的硫磷堆積物,CO和C3H6的起燃溫度較未再生的催化劑均降低了30 ℃[77]。Christou等用草酸在50 ℃的條件下清洗三元催化轉換器(TWC)表面,該方法可以使因硫磷中毒而封閉的孔道重新打開,使催化劑恢復一定的催化活性。與中毒催化劑相比,CO和HC的排放降低了50%,NO的排放降低了32%[78-79]。Subramanian等使用乙二胺二琥珀酸清洗TWC進行再生,該方法可以去除大部分磷、鈣、鋅和全部的硫,并且清洗劑容易降解[80]。Christou等用二氯乙烷和二氯乙酸清洗磷中毒的TWC,催化劑表面覆蓋的磷酸鹽被清洗,原本被掩蓋的Pd暴露至表面,表面探測得到的Pd含量較毒化的催化劑增加了4倍,CO和C3H6的T50均降低了50 ℃[81]。通過試劑清洗難以直接實現已經燒結的活性組分的再分散,但是使用含氯試劑清洗后,表面殘留的Cl-有助于熱再生過程中貴金屬的再分散,因此二氯乙烷和二氯乙酸是一種有潛力的清洗劑,可以用來清洗中毒物質并輔助再生[82]。
磷中毒會導致整體式DOC不可逆失活,但是目前磷中毒機理以及緩解策略的相關研究尚不系統,仍需要進一步完善,主要為以下兩個方面:①磷中毒方式未統一,試驗模擬磷中毒的方式不同,主要為浸漬中毒和含磷氣體處理,采用的中毒物質也存在差異,有磷酸、磷酸氫銨等,無法確認這些因素是否會對磷中毒機理的研究產生影響;②清洗催化劑是磷中毒后常用的再生方式,雖然可以有效去除大部分中毒組分,但難以在柴油機運行過程中實現原位操作,操作難度和成本極高,即使實現原位操作后,清洗是否會對處理系統中其他部件造成影響也是未知數。因此,催化劑的磷中毒緩解策略需要以抗磷改性為主,削弱磷與催化劑之間的相互作用,這需要深入探究磷中毒機理,為催化劑的抗磷改性提供指導。
3 硫磷復合中毒
在實際運行中,硫和磷往往同時對DOC造成毒害。Lanzerath等對DOC進行臺架試驗后發現,運行124 328 km的DOC表面同時堆積了質量分數為1.4%的磷和1.8%的硫,NO的最大轉化率由80%下降到了40%[83]。Agote-Arán等對運行288 000 km后的DOC進行取樣,發現其表面同時積累了6 065 μg/g的磷和975 μg/g的硫,CO的T20上升了22 ℃,C3H6的T90上升了24 ℃,NO的最大轉化率由63%下降到了11%[63]。表3展示了部分整體式DOC經過硫磷毒化后的活性變化。Honkanen等研究發現,在商用DOC硫中毒的情況下,CO的T90上升了12 ℃,C3H6的T90上升了20 ℃,但在磷硫同時中毒的情況下,將溫度升至300 ℃也無法完全轉化C3H6,且NO氧化能力幾乎完全喪失[25]。V?liheikki等對比了整體式Pt/SiO2-ZrO2催化劑磷中毒和復合中毒的催化性能,研究結果表明,僅磷中毒時,CO和C3H6的T50提高了30 ℃以上,而復合中毒時,催化劑的中毒效應得到了一定的緩解,CO和HC的T50均僅提升了10 ℃[84]。
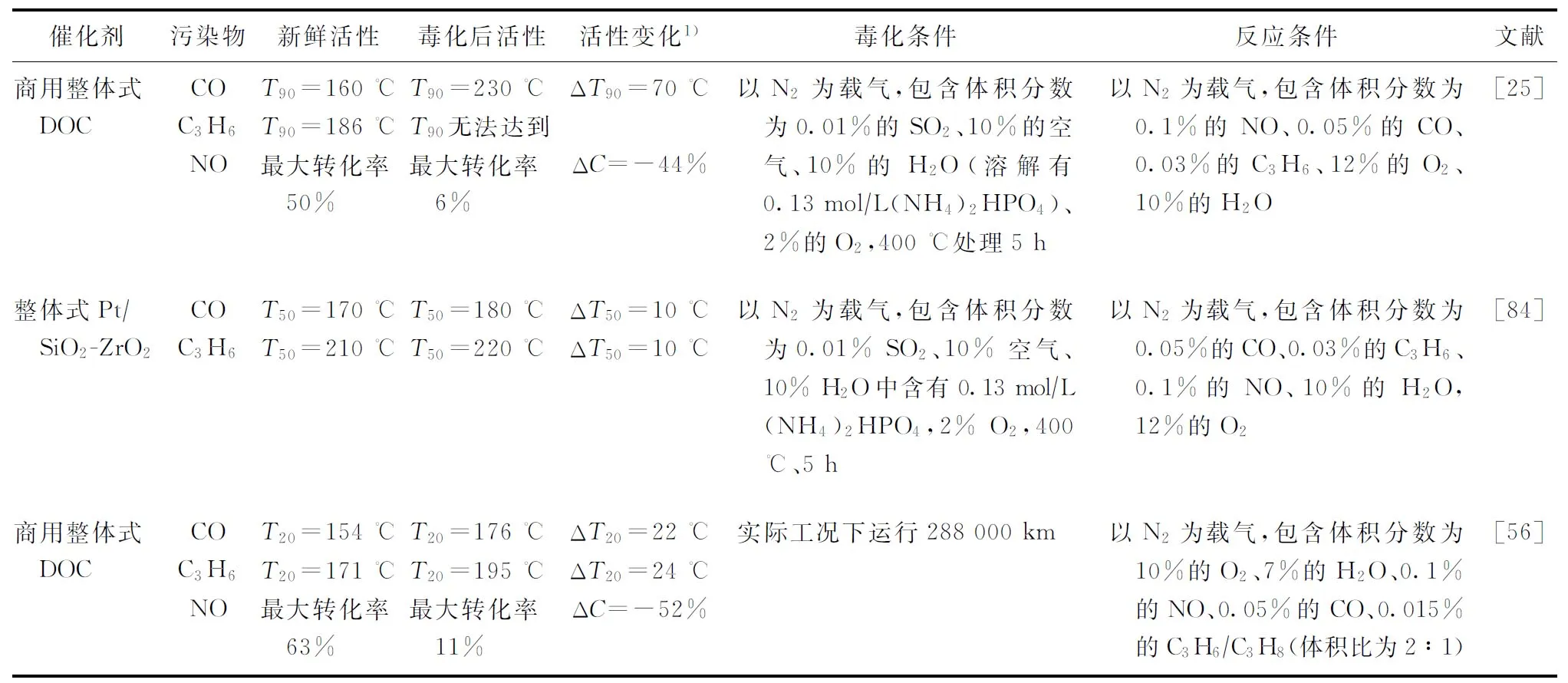
表3 催化劑硫磷中毒前后活性對比
硫磷復合中毒時,硫和磷之間會產生協同效應。Honkanen等研究發現,硫磷同時存在對Pt/Al2O3催化劑活性的影響遠高于單一組分。硫磷復合毒化的原理如圖4所示,由于磷在催化劑表面形成磷酸鋁,阻礙了吸附在Pt表面的硫物種向載體遷移,使硫堆積在Pt表面,繼而加劇活性位點Pt的失活[25]。目前硫磷中毒對催化劑結構和性能的影響研究較少,與之相比,硫磷在整體式催化劑上的分布研究更為系統、完善。整體式催化劑中,在硫磷復合中毒的情況下,磷沉積到催化劑表面后,與載體涂層的陽離子結合能力較強,會優先和載體結合形成較硫酸鹽更穩定的磷酸鹽,并且磷沉積后載體酸性增加,可削弱硫的吸附。因此,硫酸鹽在磷酸鹽沉積區域往往較少[56]。磷硫分布和催化劑涂覆方式、貴金屬涂覆比例等因素密切相關。2020年以前對整體式催化劑的研究發現(圖5),經過217 261 km運行的DOC上,硫、磷均在進氣端堆積。在涂層的深淺分布上,磷酸鹽主要堆積在表面,而硫酸鹽由于受到磷酸鹽的抑制難以在表面堆積,會向催化劑涂層的內部擴散[37,83]。這可能是由于早年DOC的活性組分(Pt、Pd)按設計含量要求在堇青石表面分段涂覆,貴金屬在進氣端涂覆量較多,而在出氣端涂覆量較少。易被硫、磷毒化的組分Pt、Pd均在進氣端聚集,導致在進氣端沉積了大量的硫磷毒物,甚至深入涂層內部。在2020年以后的研究中,硫磷在整體式催化劑上分布的研究呈現出不同的結果,研究結果顯示,磷大量沉積在進氣端,而硫在出氣端聚集。Agote-Arán等研究發現,在經過288 000 km運行的DOC上,磷質量分數為6 065 μg/g的進氣端中硫質量分數僅為975 μg/g,而在磷質量分數為3 100 μg/g的出氣端,硫質量分數上升到了1 335 μg/g[55-56,63]。在涂層深淺分布上,在磷含量較多的進氣端,磷會均勻分布在涂層內,硫由于受到磷的抑制只能在涂層表面堆積;在磷含量較低的出氣端,硫在涂層內部和外部均有分布。這可能是由于近年來涂覆技術改進,DOC的活性組分按不同比例在堇青石上分段涂覆,進氣端Pt涂覆量更高,而出氣端Pd涂覆量更高。Pt更容易受到磷的影響,這使得本身易在進氣端堆積的磷化物可以深入涂層內部,而容易硫中毒的Pd催化劑在出氣端涂覆量較多,并且進氣端涂層中分布著更易與Pt結合的磷,致使硫的分布向出氣端聚集。但是當涂層中存在助劑Ce時,硫可以突破磷酸鹽的阻礙進入涂層內部并形成硫酸鈰,毒化催化劑[85]。但目前沒有系統地探究不同DOC工藝導致DOC硫、磷中毒后呈現不同中毒狀態的具體機理,并且實際運行過程中堿金屬、灰分等因素是否也會影響硫、磷的中毒狀況也未得到系統研究。

圖4 硫磷復合毒化原理示意[25]
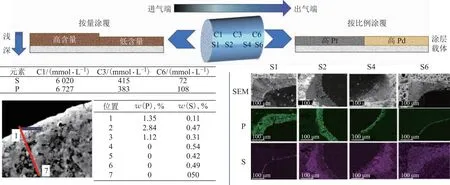
圖5 整體式DOC中毒示意(進氣端到出氣端順序分別為S1、S2、S4、S6[56],C1、C3、C6[37])
緩解硫磷復合中毒的方式與硫、磷單一中毒接近,主要是減少硫磷在催化劑上的堆積,目前相關研究也較少。V?liheikki等用酸性復合氧化物SiO2-ZrO2作為Pt載體,該催化劑磷硫復合中毒時的活性優于硫、磷單組分中毒,這是由于硫和磷在SiO2-ZrO2載體上的吸附能力接近,二者競爭相同的吸附位點,一種物質堆積的同時會抑制另一種物質的吸附,避免了硫和磷吸附在不同位點,從而加劇催化劑的毒化[58]。
實際運行過程中,DOC硫磷復合中毒最常見。但相關研究主要集中在整體式催化劑的中毒情況上,對硫磷復合中毒機理的研究較為欠缺,如磷硫復合中毒對貴金屬賦存狀態、配位特性等影響機制,在復合中毒情況下載體酸堿性對貴金屬-載體相互作用、活性中心結構等影響機制。由于對磷硫復合中毒機制的認識欠缺,因此對催化劑硫磷復合中毒緩解措施的相關研究也進展緩慢,亟待推進。
4 總結與展望
柴油車尾氣中不可避免地含有硫、磷,而處于整個后處理系統最前端的DOC首當其沖極易受到硫、磷毒化。
尾氣中硫、磷的存在會嚴重影響DOC的活性及耐久性能,而硫、磷同時存在一般會加劇催化劑中毒。硫、磷既可以改變活性組分的賦存狀態,從而直接影響催化劑本征活性;也可通過降低比表面積、堵塞孔道、覆蓋活性位點的方式影響CO、HCs等污染物的傳質過程,繼而導致催化劑失活。然而,目前關于硫磷復合中毒機制的研究較少,硫磷復合中毒對貴金屬賦存狀態、配位特性等影響機制,以及復合中毒時載體和助劑對貴金屬-載體相互作用、活性中心結構等影響機制尚不清晰,亟需進一步研究。
緩解硫磷中毒的策略主要包括提升DOC抗毒性和中毒后再生性能兩種方式。雖然目前DOC的抗硫性有所提升,但仍無法避免長時間內硫酸鹽的沉積,需進一步優化;高溫加熱或外加還原劑等手段也可有效再生硫中毒催化劑,但難以在實際應用過程中實現有效再生。目前,提升催化劑抗磷或抗硫磷性能的手段有限,且效果一般,開發出抗硫磷中毒性能優異的DOC仍是一個挑戰。由于磷酸鹽較為穩定,通過高溫加熱也難以再生,具有類似組分的三效催化劑磷中毒有效再生手段為弱酸清洗,但在DOC上是否適用,還需進一步驗證。
為滿足實際應用需求,后續的相關研究應重點關注以下幾方面:①由于實際工況面臨硫、磷、堿金屬、灰分等多污染物的復雜環境,需重點研究多污染物組分共存時,DOC材料的復合中毒失活機制;②要借助原位表征和計算等手段,深入剖析硫、磷等污染物組分對活性組分賦存狀態、配位特性等影響,為高抗毒性催化劑的設計提供理論支撐;③相較于中毒后再生,高抗毒性催化劑的設計開發是解決中毒問題的更佳策略;④可利用計算機建立催化劑中毒失活模型,將催化劑物理結構和化學狀態變化設為參數,根據以往數據,預測新研發的抗毒技術在整體式催化劑上應用后的失活曲線,以降低產業化應用的時間和成本,助力打通原理性技術到工業化應用的技術瓶頸。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
