淡水魚類環境DNA宏條形碼引物的篩選及其在千島湖的應用*
2024-01-13 07:41:38胡文靜李志力劉其根胡忠軍
湖泊科學 2024年1期
周 嚴,童 璐,胡文靜,李志力,郝 雷,劉其根,胡忠軍**
(1:上海海洋大學,農業農村部魚類營養與環境生態研究中心,上海 201306) (2:上海海洋大學,農業農村部淡水水產種質資源重點實驗室,上海 201306)
維持生物多樣性的穩定是生態系統可持續發展的基礎,由于人類的過度開發、氣候變化、環境污染、外來物種入侵等因素,全球野生動物資源量正在大幅縮減,據2022年世界自然基金會發布的地球生命力報告顯示,自1970-2018年之間,全球野生動物種群數量消亡了69%,地球生命力指數下降幅度超過一半以上[1]。截至目前,全球共有3萬多種魚類,占所有脊椎動物的一半以上,具有重要的生態效益和經濟價值[2-3]。隨著對魚類受威脅狀況了解的深入,魚類所受威脅的認知度也不斷加深,以中國為例,在最新的《中國生物多樣性紅色名錄》(脊椎動物卷)評估中受威脅的魚類比例高達22.5%,魚類保護工作的實施已迫在眉睫[4]。魚類多樣性的有效保護依賴于對現有魚類群落生態和動態的深入了解,這需要對魚類的群落結構進行全面和準確的評估。傳統的魚類多樣性評估多依靠捕撈如電捕、網捕等方式進行樣品的采集,不僅需要大量的人力和時間,而且成本也較高;同時,基于形態學的物種鑒定非常容易出錯,處在生活史早期階段的魚類鑒定尤為如此,因此魚類樣品的準確分類需要依賴專業的鑒定人員[5]。此外,基于捕獲的調查方式往往會對魚類本身造成傷害,反而使得魚類的資源量變得更少,這也違背了生物多樣性保護工作的初衷[6]。
eDNA(environmental DNA)是指從環境樣品(如土壤、水或空氣)中提取的片段DNA,不需要預先捕獲任何目標生物[7]。eDNA方法是基于直接從環境樣本提取到的生物體DNA進行序列分析,可以在不捕獲實際生物體的情況下對目標物種進行識別[8]。自2008年Ficetola首次成功將eDNA技術應用于水生生物調查以來,越來越多的人開始使用這種方法,基于eDNA的生物多樣性監測已被證明是一種高效、經濟和無創的生物監測方法[8-10]。將宏條形碼引物與高通量測序相結合,eDNA方法在魚類多樣性監測方面取得了巨大的進步,與傳統的調查方式相比較,其能夠更好地揭示物種的豐富度,已徹底改變了動植物資源的調查方式[11-13]。
目前,基于eDNA的生物多樣性監測技術仍不夠完善,需進一步開展系統的研究和驗證[14]。在技術應用方面,宏條形碼引物合適與否常被認為是影響eDNA監測結果準確性的主要因素之一[15]。不同引物對于魚類物種的覆蓋率和分辨能力有所不同,尤其是當引物對于魚類環境DNA的擴增效率比較低、存在許多非目標物種的擴增,或對不同物種的序列區分能力不足時,往往會出現假陽性或者假陰性的鑒定結果,而理想的引物應具有較高的物種覆蓋率、較強的物種分辨率、適當的條形碼長度以及更少的非特異性擴增等優勢[15-16]。因此,引物的開發或篩選是提高eDNA監測效率和可靠性的關鍵步驟[13,17]。
過去有研究者針對魚類群落開發了部分宏條形碼引物,并且成功地描述了當地的魚類多樣性[18-19]。由于很難對這些來自不同研究中的宏條形碼引物的監測效果進行比較與評估,許多研究人員僅依據個人的主觀判斷從眾多引物中挑選一對引物對魚類物種多樣性展開研究,并未深入分析該引物的特征以及其對所研究群落的適用性[20-21]。近年來,相關研究開展了eDNA宏條形碼引物的分析和測試,但僅評估了少量的引物[22],也有部分研究選取較多的引物開展了相對全面的評估[16,23],伴隨新的引物不斷被開發出來[24-25],與之前常用的宏條形碼引物相比,新開發出來的引物是否有更好的表現,仍需進一步驗證。因此,有必要對現有的宏條形碼引物進行全面評估,并探討它們在基于eDNA技術的魚類物種監測中的適用性和可靠性。
本研究結合計算機模擬PCR和野外樣品實驗驗證,比較評估已發表的基于12S、16S、COI和Cytb線粒體基因設計的27對引物,以及自行設計的2對引物,共29對引物。首先,計算機模擬PCR分析來自Mitofish數據庫魚類基因數據的物種覆蓋率和擴增長度等,接著對來自千島湖五大湖區及其支流的eDNA樣品進行宏條形碼分析以綜合評價不同宏條形碼引物優劣勢,為今后基于eDNA技術的魚類物種多樣性評估提供參考。
1 材料和方法
1.1 引物的檢索與設計
為盡可能全面地搜集有關魚類的宏條形碼引物,在科學網(www.webofscience.com)以eDNA或environmental DNA或eDNA metabarcoding為關鍵詞進行文獻檢索,共檢索到27對已應用于eDNA研究且效果較好的宏條形碼引物,包括了12S rRNA、16S rRNA、COI和Cytb基因。同時,基于千島湖歷史記錄中的魚類物種,從NCBI(https://www.ncbi.nlm.nih.gov/)和Mitofish數據庫(http://mitofish.aori.u-tokyo.ac.jp/)中下載了105種魚類的127條線粒體DNA序列,使用Primer Premier 5.0軟件[26]基于12S rRNA區域設計了2對宏條形碼引物,以上所得到的eDNA宏條形碼引物匯總如表1所示。

表1 本研究分析的22對魚類宏條形碼引物Tab.1 Summary of the 22 fish metabarcoding primer sets analyzed in this study
1.2 計算機模擬PCR
于2021年11月,從Mitofish數據庫中下載3118種魚類的線粒體DNA序列,構建了標準魚類線粒體DNA條形碼數據庫。首先,使用PrimerMiner對Mitofish數據庫中3118種魚類進行評估,以此來計算宏條形碼引物對于Mitofish數據庫中不同魚類物種的覆蓋率和分辨率[40],允許的最大錯配數為3 bp。隨后使用Tbtools對成功擴增的序列長度進行分析[41]。最后通過megaX計算擴增序列之間的平均遺傳距離[42],以評估不同宏條形碼引物所擴增魚類序列之間的差異程度。當引物擴增的物種數目大于所有引物擴增魚類物種數的平均值且平均擴增長度處于100~500 bp之間時,該引物被用于下一步實驗,以進一步評估其對于實際環境DNA樣本中魚類的監測能力。
1.3 eDNA樣品的采集與保存
千島湖位于浙江省杭州市淳安縣境內,有著良好的生態環境和豐富的魚類資源,自20世紀以來的歷史標本和網捕調查結果記錄了上百種魚類[43]。為了評估引物對于實際采集的環境DNA中魚類的監測效果,本研究對來自千島湖湖區及其支流的水環境DNA進行了驗證。在2020年12月-2021年10月,從千島湖五大湖區和14條支流共計39個站點進行了水樣的采集。通過玻璃采水器,在每個采樣點水下約0.5、5和10 m的位置收集3個2 L的水樣,將水樣混合均勻后放置在冰上運回上海海洋大學千島湖基地實驗室,在12 h內全部過濾。抽濾過程中所涉及的鑷子、過濾器均提前置于10%的次氯酸鈉溶液中浸泡10 min,并使用純水沖洗干凈。每次過濾前均設置2 L無菌水作為陰性對照,使用孔徑0.22 μm的混合纖維素濾膜(生工生物)對采集的水樣進行過濾,過濾完后將濾膜對折,放置于2 mL離心管中,采用干冰冷凍保存。
1.4 傳統魚類資源調查
于2020年9月-2021年11月對千島湖五大湖區及其主要支流開展了野外調查,在千島湖湖區及河湖交匯處的淌水區表層、5 m水層、10 m水層分別放置多網目單層刺網1套,在沿岸帶放置多網目單層刺網1套、定置串聯倒須籠網2個。支流主要依靠定置串聯倒須籠網的方式進行魚類多樣性調查,在每個采樣點放置定置串聯倒須籠網2個。傍晚日落前1 h左右放網,其在水中浸泡約12 h后,次日清晨收網。收集所有魚類,用低溫保溫箱將魚類運回上海海洋大學千島湖基地實驗室,進行物種鑒定和形態學數據的測量。
1.5 eDNA的提取、PCR擴增和測序
樣品運輸至實驗室后,采用Fast DNA Spin Kit(MPBio)試劑盒并參照說明書進行總DNA的提取。提取后的DNA經1%瓊脂糖凝膠電泳檢測,并用NanoDrop 2000質檢合格后于-20℃保存備用。對于計算機模擬PCR中擴增長度在100~500 bp且擴增出的物種數在所有引物擴增魚類物種數的平均值之上的引物進行下一步實驗,以原始設計論文中使用的Tm值為中心,在10~20℃范圍內的不同溫度梯度下,使用通用程序執行8~12個重復PCR,確定每個引物組的最佳退火溫度。針對每一對引物,均進行至少兩次梯度PCR實驗,以確保擴增效果的一致性。對于每個引物組,在明確最適退火溫度后,使用已提取好的21個eDNA樣本和3個陰性對照進行PCR擴增,每一次PCR均為3個重復,以確保PCR產物的量足以進行測序。每次PCR反應的總體積為25 μL,其中模版DNA 1 μL、正反引物各1 μL(10 μmol)、Ultra HiFidelity PCR Kit II(天根生物)12.5 μL,ddH2O 9.5 μL。PCR反應程序為:98℃ 30 s;98℃ 10 s,退火溫度(Tm)20 s,72℃ 20 s,35個循環;72℃ 5 min。隨后通過1%瓊脂糖凝膠電泳對PCR產物進行檢測,確保檢測結果均為一條單一明亮的條帶,將每一條帶進行切膠,純化回收,并用NanoDrop 2000分光光度計(Thermo Scientific)測定DNA濃度,將每組引物擴增純化后的21個PCR產物進行等量混合,利用Illumina公司的Miseq PE300平臺進行測序,進一步的文庫構建和上機測序由上海美吉生物技術有限公司進行。
1.6 測序數據的生物信息學處理
基于fastp[44](https://github.com/OpenGene/fastp,version 0.20.0)軟件去除引物接頭序列,并對原始測序序列進行質控,過濾掉低質量的Reads。采用FLASH[45](http://www.cbcb.umd.edu/software/flash,version 1.2.7)軟件根據Reads間的重疊關系,對嚴格過濾后的Reads進行拼接。使用軟件USEARCH(v7.0.1090)根據97%的相似度將序列聚類為可操作分類單元(OTU)[46]。為了解每個OTU所對應的物種,采用BLAST算法對OTU代表序列進行分類學比對,參考數據庫為Nt數據庫(https://www.ncbi.nlm.nih.gov/),將序列一致性≥97%作為閾值,對OTU代表序列進行注釋。使用Qiime軟件基于bray_curtis距離算法計算beta多樣性距離矩陣[47],使用R語言(version 3.3.1)vegan軟件包進行NMDS分析和作圖[48]。
2 結果
2.1 計算機模擬PCR分析
本研究通過計算機模擬PCR對來自Mitofish數據庫中的3118種魚類序列進行擴增,不同宏條形碼引物對于不同魚類物種的覆蓋度和分辨率分析結果如圖1所示。使用29對引物所擴增出的平均魚類物種數為2523種,引物12S-V52擴增出的魚類物種數最多,一共擴增出了3113種魚類,引物GFCC擴增的魚類物種數最少,僅擴增出了429種魚類。對擴增出的魚類物種作進一步分析,平均每對引物能夠準確鑒定的物種數為2144種,引物16SAR/BR能夠準確鑒定的魚類物種數最多,為2782種,引物GFCC能夠準確鑒定的物種數最少,為336種。

圖1 不同引物能夠擴增出的物種數和能夠明確鑒定出的魚類物種數量Fig.1 Number of species that can be amplified and number of fish species that can be unambiguously identified by different primers
通過對不同宏條形碼引物擴增出的魚類序列長度作進一步分析,大多數宏條形碼引物的擴增長度處于100~500 bp之間。不同eDNA宏條形碼引物擴增的平均條形碼長度為305 bp,其中FishF1/R1擴增出了最長的長度,為655 bp,引物Teleo擴增出了最短的長度,為63 bp(圖2)。

圖2 基于計算機模擬PCR結果計算出的每對引物平均擴增長度(虛線表示用于下一步實驗驗證時擴增片段長度的閾值)Fig.2 Average amplification length per primer pair calculated based in silico PCR results
2.2 遺傳距離分析
基于不同引物擴增出的魚類物種,計算了不同物種之間的平均遺傳距離。29對引物擴增的魚類物種間遺傳距離的平均值為19.02%±3.52%(圖3),引物ChordB擴增出的魚類序列之間平均遺傳距離最大,其次是Fish16S和16SFishS,而引物L2513擴增出的魚類序列之間平均遺傳距離最小。
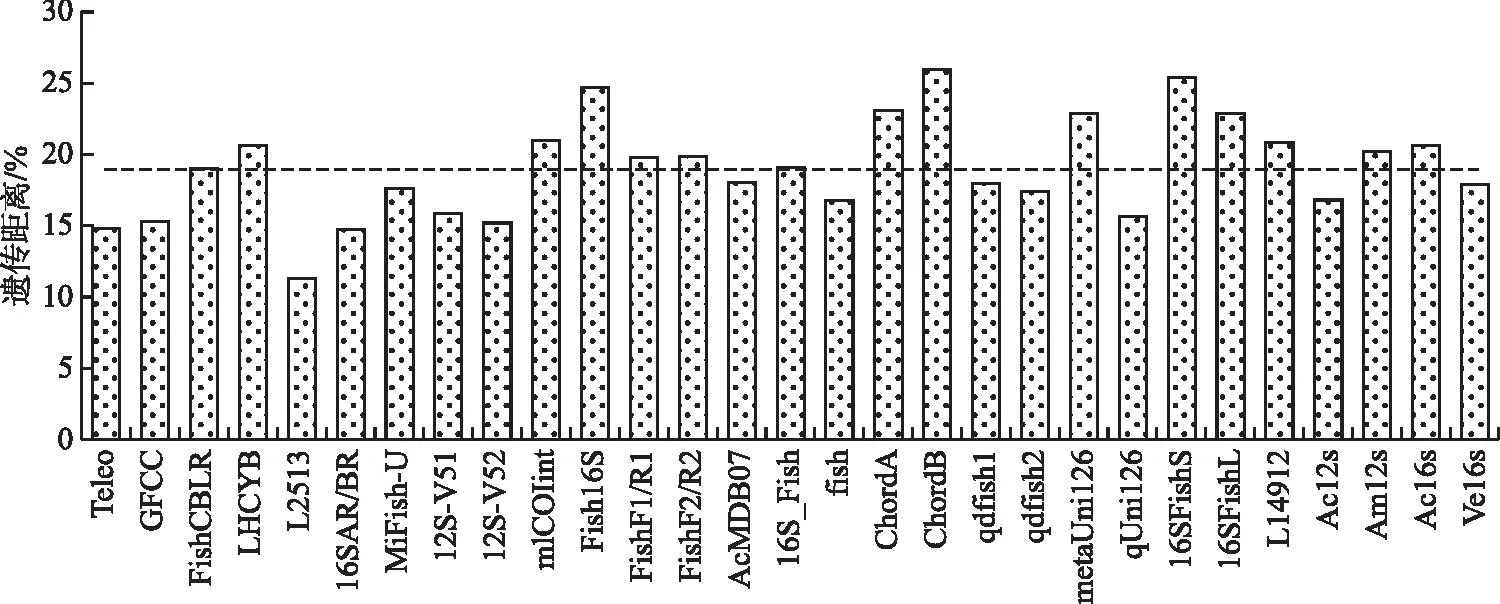
圖3 不同引物擴增出的魚類序列之間的遺傳距離(虛線表示平均值)Fig.3 Genetic distance between fish sequences amplified by different primers
2.3 高通量測序結果
基于計算機模擬PCR的結果,有17對引物成功擴增的物種數以及擴增片段長度處于符合的范圍內而被用于下一步分析。通過對已提取的eDNA樣本進行PCR擴增和上機測序,經過質控、拼接、聚類以及數據庫比對,17對引物中有15對被成功擴增并得到了與之相關的測序結果,另外2對引物由于擴增的序列沒有匹配到具有≥97%相似性的魚類序列而被刪去。15對引物所構建的測序文庫共獲得有效拼接序列961740條,有效拼接序列最多的為Ac16s,共獲得127385條序列,最少的為Ac12s,共獲得31255條序列。從物種稀釋曲線圖(圖4)可知,隨著測序量的增加,曲線逐漸趨向于平緩,由此可以看出測序深度足以覆蓋樣品中所有的物種,可以用于后續分析。
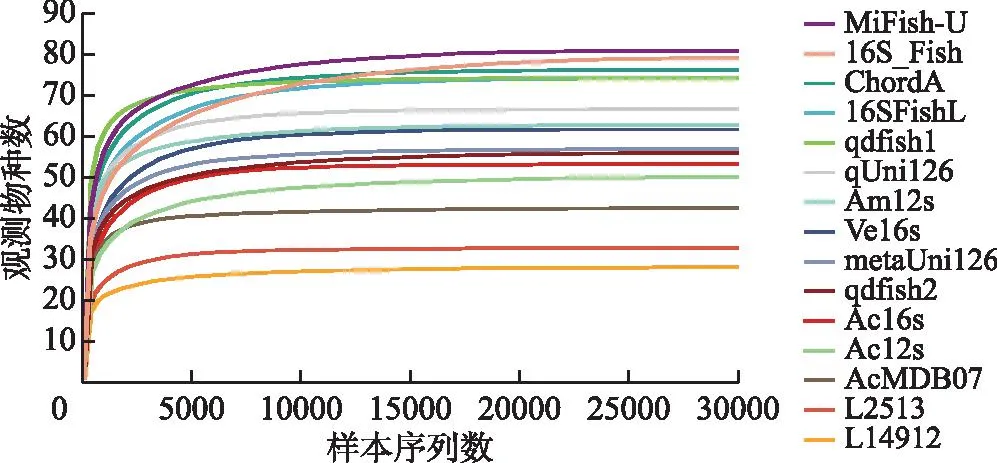
圖4 使用不同引物所得到的物種稀釋曲線Fig.4 Rarefaction curves of the observed species in different primers
2.4 魚類物種組成
不同宏條形碼引物所監測到的魚類物種數量如圖5所示,平均每對引物監測出的魚類物種數為60種,引物MiFish-U、16S_Fish、ChordA、16SFishL、qdfish1監測到的魚類物種數相對較多,而其他引物監測到的物種數相對較少(如L14912只擴增出了28種魚類)。將不同宏條形碼引物監測到的魚類物種與傳統魚類資源調查結果進行對比,盡管監測到的物種數排名前四的引物仍為MiFish-U、16S_Fish、ChordA、16SFishL,但監測到物種數最多的宏條形碼引物是ChordA(58種),其次是16SFishL(57種),然后是引物16S_Fish和MiFish-U。
基于不同的宏條形碼引物所獲得測序結果,選取豐度排名前50的魚類物種,繪制了魚類物種組成差異圖。如圖6所示,豐度排名前50的魚類中,16SFishL、ChordA、Mifish-U、qdfish1監測的物種數最多,均監測到了40種以上的魚類;而引物L14912和L2513監測到的物種數最少,僅為19種。不同的引物既顯示出了各自所具備的特性也顯示出了不足之處,一方面,本研究中所用到的引物對于大部分魚類物種均能夠監測到,另一方面,也有一些物種僅僅只被一對引物或個別引物所監測到。
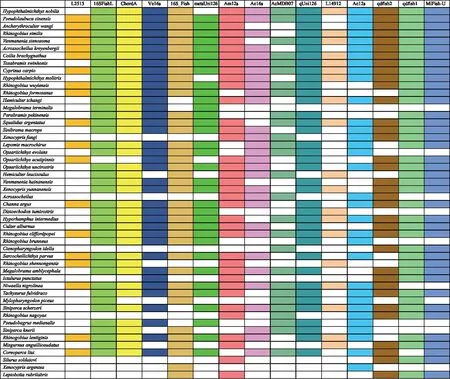
圖6 不同引物監測到的魚類物種差異圖(豐度排名前50)Fig.6 Map of differences in fish species monitored by different primers (top 50 in abundance)
2.5 群落結構組成
基于不同引物所獲得的高通量測序結果,繪制了不同物種的序列分布柱狀圖(圖7)。引物L2513、Ve16s和qUni126擴增出的魚類序列數只占總體序列數的50%左右,引物Ac12s擴增出的魚類序列數占所有魚類序列數的70%~80%之間,引物metaUni126、Am12s、qdfish2和qdfish1擴增出的魚類序列數在80%~90%之間,如Am12s(84.42%);其余6對引物擴增的魚類序列數占比都大于90%。在大部分引物所擴增出序列中,魚類的序列都是最多的,但也有一部分引物的特異性不是很好,除了擴增出魚類的DNA序列之外,也擴增出了一些其他類群物種的序列,如鳥類、爬行類、兩棲類等。

圖7 不同引物基于體外eDNA宏條形碼分析所得的序列分布Fig.7 Sequence distribution of different primers based on in vitro eDNA metabarcoding analysis
將針對不同目的基因的宏條形碼引物分為3類進行NMDS分析,結果如圖8所示。NMDS分析計算出stress值為0.1,小于0.2,表明分析結果具有一定的可靠性,可以反映出不同引物監測到的魚類物種間的差異程度。基于相同的eDNA樣本,盡管不同的引物產生了不同的魚類多樣性特征,但不同組之間沒有顯著的差異。
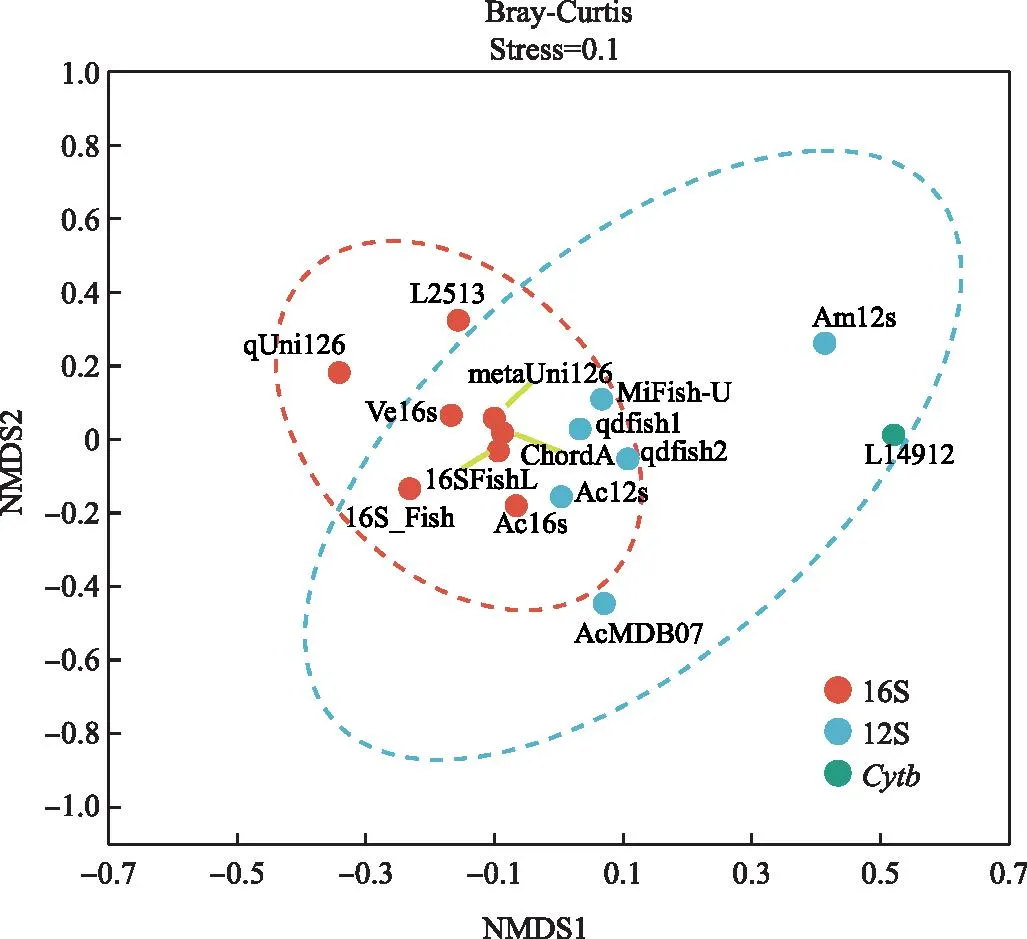
圖8 魚類群落結構的NMDS分析Fig.8 NMDS analysis of fish community structure
3 討論
3.1 不同引物對于魚類的覆蓋度和分辨能力分析
對不同魚類物種有著更廣泛的覆蓋范圍并且能夠有更大的區分程度是使用eDNA宏條形碼對魚類物種多樣性進行監測的先決條件。通過計算機模擬PCR發現,不同的宏條形碼引物對魚類的擴增效率和分辨能力也有所不同。總的來說,COI和Cytb基因對于魚類物種的分辨能力并不差,除了引物GFCC,其他引物擴增出的魚類序列之間的遺傳距離均處于平均值以上。此外,COI基因參考數據庫所覆蓋的物種也更加廣泛[49]。然而,基于12S和16S基因設計的宏條形碼引物比基于COI和Cytb的宏條形碼引物所擴增出的魚類物種數更多,因而能夠區分出的魚類物種數也更多。對此,以往的研究者也有著相似的發現,陳治等[50]通過對海南島淡水魚類的DNA條形碼研究發現,對于魚類物種判別能力最高的目標基因為COI,其次為16S和12S。但在實際的魚類環境DNA研究中卻很少用COI作為目標基因,其最大原因在于COI很難設計出DNA宏條形碼通用引物[18],且也有研究表明,對水生動物DNA濃度低的水環境而言,靶向于魚類COI基因的引物比12S表現較差[51],限制了COI引物在基于eDNA技術的魚類多樣性監測中的應用。
在模擬PCR后對其中的17對宏條形碼引物作了進一步驗證,分析結果顯示,盡管17對引物在模擬PCR中的分析結果均顯示良好,但在實際的擴增過程中,結果卻各有不同。17對引物中有2對因未能夠成功擴增出魚類的序列而被刪除,其余15對宏條形碼引物最多的監測到了81種魚類,最少的僅監測到了28種魚類。由此可以看出,模擬PCR可以幫助我們增加對于引物本身的了解,但模擬PCR仍需要與實際的宏條形碼分析結合起來。監測到魚類物種數最多的5對引物中,有3對是基于16S rRNA設計的,有2對是基于12S rRNA設計的,這也表明了基于12S和16S基因都能夠設計出較好的引物以用于eDNA研究。具體來看,不同引物對于魚類的覆蓋度和分辨能力也有所不同,如MiFish-U、16S_Fish、ChordA、16SFishL和qdfish1對魚類的覆蓋度更高一些,但從不同引物對擴增的魚類物種的分辨能力來看,16SAR/BR、qUni126和Ve16s又是最好的。基于不同的宏條形碼引物所獲得測序結果,一方面不同的引物都監測到了一些相同的魚類物種,使得它們之間可以相互印證,增加了研究的可靠性;另一方面,由于不同引物具有不同的特性和優缺點,互相補充能夠最大程度地提高魚類的檢測概率,因此在今后的基于eDNA的物種多樣性研究中,推薦采用多對宏條形碼引物進行監測。
3.2 不同引物的特異性比較
對魚類序列進行特異性擴增是魚類環境DNA宏條形碼引物選擇的重點,如果對魚類物種序列的特異性不夠高,則在PCR擴增的過程中就可能過度擴增其他物種的序列,從而占用大量的測序通量并導致魚類的測序量不足,無法獲得足夠的魚類分類學信息[52]。本研究結果顯示,幾乎每一對引物都擴增出了非魚類物種的序列,甚至有個別引物擴增出來的非魚類物種的序列占比達到了所有物種的一半以上。Zhang等[16]的研究結果也發現,幾乎所有魚類宏條形碼引物都擴增出了非魚類物種的序列,這表明對于eDNA研究而言,寄希望于引物能夠完全不擴增非目標物種似乎是不太現實的。通常而言,擴增出來的非目標物種大多是人類、鳥類、哺乳類、爬行動物、兩棲動物和細菌等。由于人類活動、動物行為及水體流動等原因,eDNA中常常會涵蓋非魚類物種序列,因此在監測到魚類物種的同時,監測到以上物種似乎不足為奇。近年來,一些研究者開始嘗試使用阻斷引物以解決這一問題[53]。然而,已有研究表明,使用阻斷引物會降低eDNA宏條形碼研究所能夠監測到的目標物種的數量;據此,一些學者提出,若非目標物種的序列較低時,不建議使用阻斷引物[16,54]。因此,為了避免非特異性擴增的出現并增加目標物種的監測概率:一方面需要充分了解目標分類群的DNA序列特征,選擇適當的引物和擴增條件以最大程度地減少非目標序列的擴增;另一方面,對eDNA研究中難免會出現擴增出非目標物種這種情形,增加測序深度或許會是一種比較可行的解決辦法。
3.3 不同引物擴增長度分析
由于eDNA數量少和易降解的特性,擴增片段長度較短的引物通常被認為可以提供更大的擴增成功率[55]。有研究者曾對水中的eDNA大小分布進行了研究,結果表明較短的線粒體DNA片段比長的DNA片段更為豐富,使用擴增子較短的引物可以獲得更大的檢測率[24,56]。但也有研究表明,較長的DNA片段能夠提供更多有關序列的信息[57]。由于較少有研究通過使用實際環境樣本系統地進行分析,往往也很難確定不同擴增子長度對于eDNA研究中物種多樣性監測結果的影響。
過去有研究者曾對22對eDNA引物進行了綜合的比較評價,結果顯示Ac12S(約388 bp)和AcMDB07(約280 bp)效果最好,監測到的物種數最多,其次是MiFish-U(約171 bp)[16]。本研究中,用于實際驗證的引物的擴增長度范圍為100~500 bp,與以往研究者的研究結果有所不同,本研究中擴增片段長度更大的引物并沒有獲得理想中最好的效果,監測到物種數最多的反而是擴增片段長度在150~250 bp之間的引物,可能的原因是,在本研究中較短的DNA片段比長的片段數量更為豐富,從而獲得了更大的擴增成功率,并監測到了更多的魚類物種。
3.4 野外樣品實驗驗證結果分析
本研究基于計算機模擬PCR篩選了17對宏條形碼引物,并對千島湖及其主要支流的eDNA樣品進行分析后發現,引物MiFish-U、16S_Fish、ChordA、16SFishL、qdfish1監測到的魚類物種數相對較多。與傳統的魚類資源調查結果相比,除了兩種方法監測到的共有種之外,每一種引物都或多或少地監測到了傳統方法未能捕獲到的魚類。這可能與以下兩方面因素有關,一方面,棲息地間的連通及水文傳輸使采集到的eDNA樣本中混入了來自其他水域的魚類DNA[58-59];另一方面,參考數據庫的不完善以及引物本身對于部分魚類的分辨率較低,導致一些物種盡管被成功擴增,但未能夠被鑒定出來[15]。
過去有研究評估了魚類分布與eDNA之間的關系,結果表明eDNA通常提供了魚類大尺度空間分布和生物量的“快照”[9]。其他一些研究也表明,基于eDNA推斷的物種時空尺度分布格局會因河流和湖泊不同而異[13]。因此,eDNA在野外條件下的釋放動態、降解和擴散模式有待進一步研究。
與傳統方法相比,eDNA方法可以在不捕獲目標物種的前提下進行生物多樣性監測,極大地改變了動植物調查的方式[13]。而無論選擇哪一種宏條形碼引物,為實現生物的準確分類鑒定,可靠且完備的參考數據庫是重中之重。公共數據庫因為結合了數千項研究的成果,產生了大量的數據,而這些數據無法臨時獲得,因此當前的eDNA研究多依賴于與公共數據庫(如GenBank或Mitofish)進行比對,從而實現物種的進一步確認。有研究表明,對于動物而言,GenBank中錯誤標記序列的比例非常低(在目水平上為0.05%,在屬水平上<1%)[60],這說明它具備較高的可靠性,可以作為了解生物多樣性的一大數據來源。然而,公共數據庫中的序列多來自分散且目的不同的研究,也沒有系統地補充和完善不同物種分類下DNA條形碼的想法和動力。由此可知,盡管公共數據庫中的序列信息仍然在不斷增長,但分類學完整的條形碼參考數據庫的建設,尤其是有關本土魚類物種序列的收集和整理仍然任重道遠。
4 結論
本研究對29對宏條形碼引物進行了篩選并在千島湖進行了初步應用,從物種覆蓋率和分辨率等角度評估了不同引物在基于eDNA的魚類多樣性研究中的適用性。本研究結果表明,eDNA技術靈敏度的確很高,可以極大地提高對于魚類的監測效率。引物的好與不好一方面取決于其本身的設計(如上下游引物的長度、退火溫度),另外一個方面也取決于數據庫的完備性。因此,對引物進行先驗性評估,然后再用于全面的生物多樣性監測十分重要。由于不同引物對魚類物種的覆蓋范圍和分辨能力也會有所不同,在這種情況下,通過使用多個引物來增加對于目標物種的覆蓋度和檢測概率,不失為一種比較好的解決方案。
猜你喜歡
英語世界(2023年10期)2023-11-17 09:18:18
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
汽車觀察(2018年10期)2018-11-06 07:05:26
財經(2017年2期)2017-03-10 14:35:35
財經(2016年15期)2016-06-03 07:38:02
財經(2016年3期)2016-03-07 07:44:46
