鈷酞菁/多孔碳氣凝膠復合結構用于高效電催化CO2 還原
2024-01-06 14:26:32龔善和呂曉萌馮玉祥黃春霞朱桂生
煤炭與化工 2023年11期
關鍵詞:催化劑
周 暾,龔善和,呂曉萌,馮玉祥,黃春霞,朱桂生
(1.江蘇索普化工股份有限公司,江蘇 鎮江 212006;2.江蘇大學環境與安全工程學院 江蘇 鎮江 212013;3.江蘇大學化學化工學院,江蘇 鎮江 212013;4.江蘇索普聚酯科技有限公司,江蘇 鎮江 212000)
0 引言
隨著全球工業化的進程,過量化石燃料轉化成CO2,并排放于空氣當中,引起了嚴重的溫室效應,引發了一系列的環境問題。目前,日益嚴重的環境問題和全球能源緊缺問題已經引起了全世界的重點關注,然而,僅憑植物光合作用難以平衡環境中的碳循環,因此迫切需要尋找一種類“自然界碳循環”的方式,來協助這一問題的解決。通過可再生能源轉換(如,風能、光能、潮汐能等)的電能,將CO2電化學還原成化學品和燃料,實現可再生能源與化學能源的耦合,從而減少溫室氣體排放,是實現碳中和的新途徑。其中,CO 作為簡單的兩電子轉移的產物,已被證明是一種具有實現工業化潛力的產物。因為其本身可直接利用,或再加工生產,以及電化學過程中令人滿意的生產率和低電能成本。然而,要實現CO2RR 工業化,仍然受到具有穩定C=O 鍵的CO2分子的高活化能壘(806 kJ·mol-1),以及CO2分子在水性電解質中的低溶解度的影響。此外,競爭性析氫反應(HER)也對提高還原產物的法拉第效率(FE)提出了巨大挑戰。因此,開發高活性、高選擇性和高穩定性的電催化CO2還原催化劑仍然是近期研究的熱點問題。
1 存在的問題
過渡金屬配合物(主要包括大環配體(卟啉、酞菁、環胺等)),多吡啶配體(聯吡啶、三聯吡啶等),鉗形配體(膦等)和卡賓配體等大環金屬分子)在均相電催化CO2還原轉化為CO 領域中的應用逐漸引起了廣泛的關注。過渡金屬的d 軌道具備多價氧化還原能力,可滿足涉及多個質子耦合電子轉移步驟的電催化CO2還原的要求,又可充當多電子反應金屬還原中心。但傳統的均相催化劑,存在:①溶解性差;②原子利用率低;③電子傳輸緩慢;④回收利用困難;⑤形成團聚而失活等問題。借助導電材料為基底,均相催化劑為催化單元,構建的多相分子催化劑,能有效解決上述存在的問題。例如,以CoPc 為催化單元,構建的Co 基多相分子催化劑,在電催化CO2還原中,表現出優異的性能。
但目前優化Co 基多相分子催化劑的工作主要集中于酞菁環外功能化修飾對位點電催化性能影響的研究,忽略了基底的孔隙結構對位點活性的影響。然而,分級多孔的結構對促進位點周圍CO2傳質,提升位點活性,是十分有效且重要的。因此,優化基底孔隙結構,以實現對催化單元微環境的優化,促進位點周圍CO2的傳質和產物的擴散,對構建高活性、穩定性的催化劑是有必要的。
碳基材料因其豐富的微觀結構和形貌、高固有電導率、優異的穩定性、重量輕和低成本而引人注目,已被廣泛用作儲能材料、催化劑、催化載體、化學吸附劑、熱絕緣體和隔音材料。其中,碳氣凝膠(CA)因其特殊性質而受到更多關注,包括高比表面積、低密度、豐富的孔隙、可控的結構、紋理、良好的導電性和化學性質。其三維(3D)分級多孔網絡骨架有利于客體離子/分子移動到內部結構中,加速催化位點和反應相關物質之間的接觸,以促進反應。
借助碳氣凝膠特殊的多級孔特性,可優化催化單元周圍CO2傳質,促進CO2與位點接觸;同時,碳氣凝良好的導電特性,將優化催化劑導電能力,促進電子界面傳導,加快電催化反應,提升基于CoPc 為催化單元的催化劑性能。通過水熱合成反應的高溫、高壓環境,合成了氮摻雜的“類珊瑚狀”多孔碳氣凝膠。通過非共價鍵固定的方法,將CoPc 穩定于氣凝膠骨架的表面,以實現活性位點和導電碳骨架穩定結合。N-CA 的特殊的多級孔結構促進了Co 位點的暴露,同時,加快位點周圍CO2傳質。因此,CoPc@N-CA 展現出的優異電催化CO2還原性能,顯著高于基于商業CB 而設計的CoPc@CB 催化劑。CoPc@N-CA 陰極催化劑,在較寬的電勢窗口(-0.5~-1.0 V vs.RHE)下,對CO的選擇性>80%,最高為99.05%,在-0.85 V vs.RHE 電位下,對CO 的部分電流密度達到14.40 mA·cm-2。
2 實驗部分
2.1 實驗藥品
乙醇(CH3CH2OH,≥99.7%)、苯酚(C6H6O,≥99.5%)、甲醛(HCHO,37%)、三聚氰胺(C3H6N6,99%)、乙酸(CH3COOH,99.7%)、N,N-二甲基甲酰胺(DMF,≥99.5%)、碳酸氫鉀(KHCO3,≥99.9%)、商品炭黑(CB)均購自國藥化學試劑有限公司。鈷酞菁(CoPc,≥96%),高純氮氣(N2,99.99%)、高純二氧化碳氣體(CO2,99.999%)和高純氬氣(Ar,99.999%)購自江蘇索普氣體有限公司(中國,江蘇)。
2.2 材料制備
2.2.1 合成氮摻雜的多孔碳氣凝膠(N-CA)
將1.882 g C6H6O 和3 mL HCHO 加入到30 mL含有0.6 mL CH3COOH 的去離子水中,然后在70℃下連續攪拌1 h。將含有0.4 mL CH3COOH、0.3 g C3H6N6和537 μL HCHO 的20 mL 去離子水加入到上述溶液中(固體前驅體預溶解后再加入),然后連續攪拌30 min。
將混合溶液放入100 mL 聚四氟乙烯高壓釜中,在160 ℃加熱16 h,得到水凝膠。將水凝膠浸入丙酮中24 h 以除去不穩定組分,然后,所得樣品通過冷凍干燥處理獲得干凝膠。所得干凝膠在N2氣氛下,800 ℃碳化3 h(升溫速率為2 ℃·min-1),然后自然冷卻至室溫得到N-CA。
2.2.2 合成酞菁鈷/氮摻雜的多孔碳氣凝膠(CoPc@N-CA)將50 mg N-CA 加入50 mL DMF 溶液中,連續超聲30 min 得到N-CA-DMF 懸濁液,標記為A 溶液。將5 mg CoPc 加入50 mL DMF 溶液中,連續超聲30 min 得到CoPc-DMF 懸濁液,標記為B 溶液。在劇烈攪拌下,將B 溶液將入到A 溶液中,并持續攪拌24 h。真空抽濾上述懸濁液,再通過DMF清洗幾次后置于60 ℃烘箱,烘干,得到CoPc@N-CA 樣品。
2.2.3 合成酞菁鈷/碳黑(CoPc@CB)
CoPc@CB 合成方法與CoPc@N-CA 相似,除了基底N-CA 被CB 替換。
2.3 電極制備
將6mgCoPc@N-CA 或CoPc@CB,3mLCH3CH2OH 和30 μL Nafion(5 wt%)溶液混合,并超聲處理30 min,以獲得催化劑油墨。將催化劑油墨溶液滴涂到碳紙(雙面涂覆,Toray,TGP-H-60,20%PTFE)的表面上,涂覆面積為1 cm,并干燥以獲得工作電極(負載量為1 mg·cm-2)。
2.4 電化學測試
采用三電極系統于H-cell 中測試材料的性能,電信號收集采用電化學工作站(上海,CHI1140c)。置于陰陽極的電極液為0.5 M KHCO3電解質(pH=7.3),H-cell 通過Nafion-117 膜分離,以防止產物的交匯。選擇銀/ 氯化銀電極(3.5 M KCl)作為參比電極,鉑(Pt)電極用作對電極。根據反應的幾何面積(1 cm2)計算電流密度。所有測得的電位通過以下公式(能斯特方程,25 ℃)轉換為可逆氫電極(vs.RHE)。
式中:nx為產生一個產物分子所需的電子數(H,CO=2);F 為法拉第常數(96 485 C mol-1);為CO2鼓泡的流速;υ 為由GC 電流信號的峰面積計算出的氣體產物的體積比,為298.15 K;Itotal:穩態測試電流(C/s)。
實驗中施加不同的電壓,進行恒電壓測試。通過配備有熱導檢測器(TCD)和火焰離子化檢測器(FID)的在線氣相色譜(Online-GC)分析氣體產物,使用DMSO 作為內標,通過1H NMR 核磁檢測液體產物。CO2氣體的流速由質量流量控制器控制在20 sccm。H 型電解池的陰極區加入轉子,攪拌速度為400 r/min,以減少傳質阻力并加速電極表面上產生的氣泡的溢出。線性掃描伏安法(LSV)測試在掃描速率為5 m·V s-1下進行。電化學活性表面積(ECSA)和電化學阻抗譜(EIS)實驗在玻碳電極上進行,玻碳電極的反應面積為0.197 cm2,催化劑涂覆量為0.15 mg·cm-2。在電壓區間為0~0.3 V vs.RHE 下,進行ECSA 的測試實驗。EIS 實驗在0.5~200 000 Hz 的測試頻率下進行,測試電位設定為-0.60 V vs.RHE。
2.5 材料表征
用配備有單色Cu Kα 輻射(λ=1.541 78A)的X 射線衍射儀(XRD,德國布魯克AXS 公司)收集樣品的粉末XRD 譜圖。拉曼(Raman)光譜由Thermo Electron Corporation DXR 拉曼光譜儀(USA)使用500 nm 激光源獲得。X 射線光電子能譜(XPS)光譜由ESCA PHI500 光譜儀檢測。場發射掃描電子顯微鏡(FESEM)圖像是用配備有能量色散X 射線光譜(EDS)的場發射掃描電子顯微分析儀(日立,Regulus-8100,日本)進行測試的。透射電子顯微鏡(TEM,日本,JEOL-2100F)在100 kV 下操作。400 MHz 核磁共振1H 譜(BRUKER AVANCE 400)來確定液體產物成分。
3 結果與討論
3.1 材料形貌表征
以三聚氰胺(M)、苯酚(P)和甲醛(F)為原料合成干凝膠。首先,在酸性條件下,P 和F 通過水熱法發生縮聚反應形成PF 聚合物。另一個縮聚反應發生在M 和F 之間,以形成MF 聚合物,MF 和PF 的羥甲基之間的進一步縮聚反應形成亞甲基醚橋連聚合物,后者可以共縮合以形成由支化聚合物種類組成的小球狀單元。進一步的水熱條件下,交錯分子的端基連續生長以形成濕凝膠的三維(3D)聚合物網絡結構。通過冷凍干燥處理濕凝膠以獲得干凝膠,干凝膠進一步碳化以形成N-CA。通過N-CA 上富電子N 位點作為軸向配體或π-π相互作用,將CoPc 錨定在N-CA 表面,形成CoPc@N-CA 樣品。CoPc 通過π-π 相互作用穩定在CB 表面,形成對比樣CoPc@CB 材料。
CoPc@N-CA 材料合成機理如圖1 所示。

圖1 CoPc@N-CA 的合成機理圖Fig.1 Synthesis mechanism diagram of CoPc@N-CA
以三聚氰胺(M)、苯酚(P)和甲醛(F)為原料合成干凝膠。首先,在酸性條件下,P 和F 通過水熱法發生縮聚反應形成PF 聚合物。另一個縮聚反應發生在M 和F 之間,以形成MF 聚合物,MF 和PF 的羥甲基之間的進一步縮聚反應形成亞甲基醚橋連聚合物,后者可以共縮合以形成由支化聚合物種類組成的小球狀單元。進一步的水熱條件下,交錯分子的端基連續生長以形成濕凝膠的三維(3D)聚合物網絡結構。通過冷凍干燥處理濕凝膠以獲得干凝膠,干凝膠進一步碳化以形成N-CA。通過N-CA 上富電子N 位點作為軸向配體或π-π相互作用,將CoPc 錨定在N-CA 表面,形成CoPc@N-CA 樣品。CoPc 通過π-π 相互作用穩定在CB 表面,形成對比樣CoPc@CB 材料。
CoPc@N-CA 的電子顯微鏡圖如圖2 所示。

圖2 CoPc@N-CA 的電子顯微鏡圖Fig.2 Electron microscope images of CoPc@N-CA
在圖2a 中,CoPc@N-CA 的場發射掃描電子顯微鏡(FESEM)圖像顯示了具有富孔結構的3D 交聯“類珊瑚狀”納米氣凝膠網絡形態,其保留了N-CA 的原始形態。其中納米交聯單元的尺寸相互連接形成氣凝膠網絡結構,以及大孔互聯的結構。這種特殊的結構將有助于CO2或CO2RR 相關物種在CO 位點周圍的大量運輸,以提高CO2RR 的活性。在CoPc@N-CA 中沒有發現CoPc 晶體殘留物,這意味著CoPc 均勻地錨定在N-CA 的表面上。CoPc@N-CA 的透射電子顯微鏡(TEM)圖像清楚地揭示了多孔無序納米網絡結構(圖2b)。對應放大的TEM(圖像(圖2c)未發現聚CoPc 的存在,說明CoPc 均勻分散在N-CA 的表面。
CoPc@CB 的電子顯微鏡圖如圖3 所示。

圖3 CoPc@CB 的電子顯微鏡圖Fig.3 Electron microscope images of CoPc@CB
使用商業CB 作為碳載體合成的CoPc@CB,通過SEM 和TEM 觀察CoPc@CB 的形貌(圖3a-b),其僅顯示與CB 相似的尺寸不均勻的大塊狀結構,與CoPc@N-CA 的“類珊瑚狀”形成鮮明的對比。同時,CoPc@CB 放大的TEM 圖像同樣未發現聚CoPc 晶體的存在(圖3c),說明CoPc 均勻負載于CB 的表面,與CoPc@N-CA 中觀察的結構類似。
CoPc@N-CA 的形貌測試圖如圖4 所示。

圖4 CoPc@N-CA 的形貌測試圖Fig.4 Morphology test images of CoPc@N-CA
CoPc@N-CA 的SEM 圖像(圖4a)和對應的能量色散X 射線光譜(EDS)元素分析圖像揭示C(圖4b)、N(圖4c)和Co(圖4d)元素均勻分布在3D 多孔納米網絡上,與上述分析結果一致,說明Co 位點均勻分散于CoPc@N-CA 中。
3.2 結構表征
CoPc@CB 和CoPc@N-CA 的表征測試圖如圖5所示。
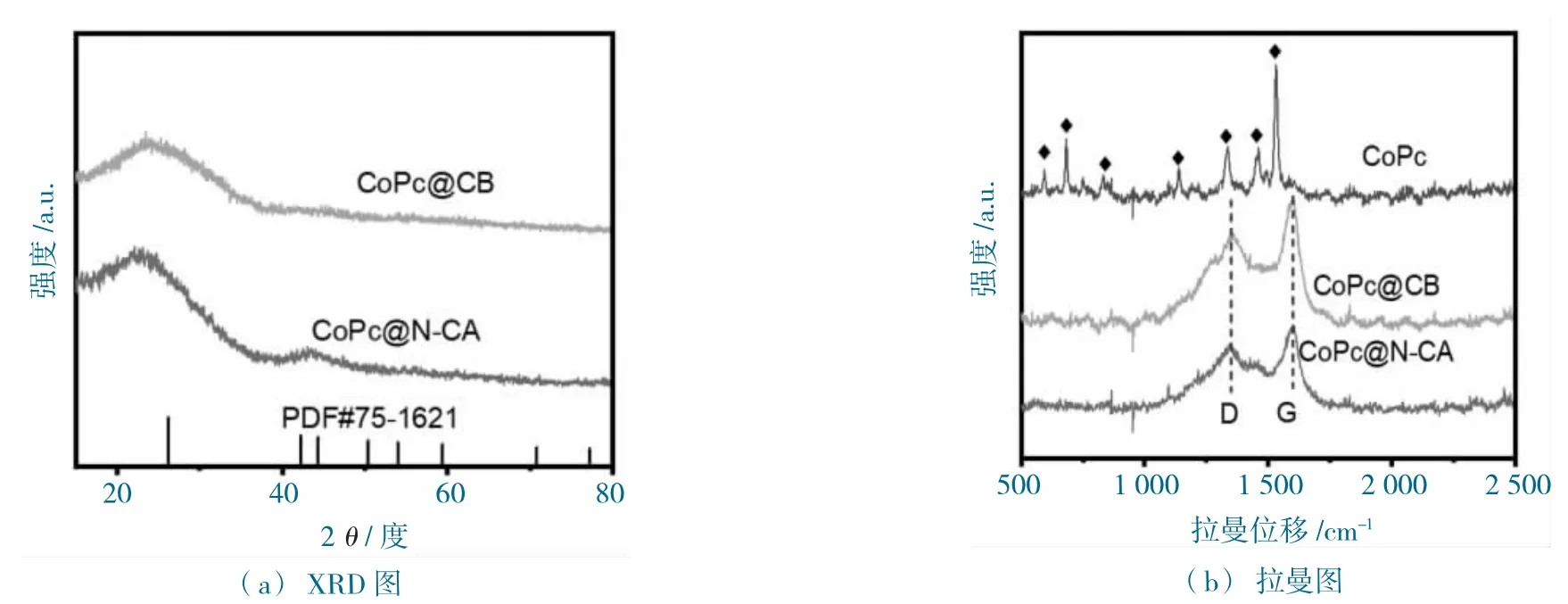
圖5 CoPc@CB 和CoPc@N-CA 的表征測試圖Fig.5 Characterization test diagram of CoPc@N-CA and CoPc@CB
由CoPc@N-CA 和CoPc@CB 的粉末X 射線衍射(XRD)圖分析發現,在23.4 和44.4 處出現2個寬峰,分別屬于石墨碳的(002)和(100)無定型晶面(PDF#75-1621,圖5a),未發現CoPc 的特征晶體峰,說明CoPc 均勻分散于基底中,這與電鏡表征的結果一致。進一步的材料Raman 光譜測試發現,商業的CoPc 在500~1 800 cm-1,顯示一系列特征譜峰(圖5b),而CoPc@N-CA 和CoPc@CB僅顯示G 帶(1 585 cm-1)和D 帶(1 351 cm-1)2 個肩峰,對應于無缺陷的sp2 碳結構和具有邊緣平面缺陷的石墨碳,未發現CoPc 的特性譜峰。
CoPc@CB 和CoPc@N-CA 的XPS 測試圖如圖6所示。
樣品中元素信息和價態通過X 射線光電子能譜(XPS)來分析(圖6a)。元素C、N、Co 存在于 CoPc@N-CA 和 CoPc@CB 中,其 中,CoPc@N-CA 中Co 的含量為0.3 at%,而CoPc@CB中Co 的含量為0.29 at%(圖6b)。
樣品中C 的主峰被矯正為284.8 eV(圖6c-d)。CoPc@N-CA 高分辨率XPS N 1s 分析發現,Co-N(399.69 eV)物種的出現,說明CoPc 的存在(圖6e)。
同時,CoPc@CB 的高分辨率XPS N 1s 分析中,也同樣發現Co-N(400.61 eV)單元的存在(圖6f),證明CoPc 穩定于CB 中。相比于商業的CoPc,CoPc@CB 和CoPc@N-CA 中的Co-N 的結合能向高的能級偏移,說明酞菁環與基底之間發生了強電子相互作用,這將有助于穩定CoPc 于基底的表面,優化位點活性。
進一步分析了Co 元素在兩個催化劑中的價態,CoPc@N-CA 中Co 2p3/2的位置為780.83 eV(圖6g),CoPc@CB 中Co 2p3/2的位置為781.12 eV(圖6h),Co 結合能的位置接近Co2+2p3/2(780.84 eV),說明樣品中的Co 的價態都為+2 價。
同時,XPS 擬合發現CoPc@N-CA 和CoPc@CB擁有一樣的 Co 2p3/2半峰寬,說明 Co 在CoPc@N-CA 和CoPc@CB 中的配位環境是一樣的。
3.3 電化學性能測試
CoPc@N-CA 和CoPc@CB 在CO2飽和的0.5 M KHCO3溶液中的電化學性能圖如圖7 所示。
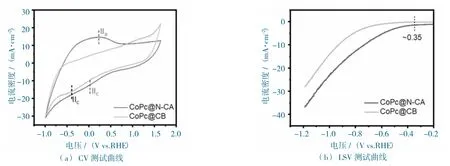
圖7 CoPc@N-CA 和CoPc@CB 在CO2 飽和的0.5 M KHCO3 溶液中的電化學性能圖Fig.7 The electrochemical performance of CoPc@N-CA and CoPc@CB at CO2-filled 0.5 M KHCO3 electrolyte.
CoPc@N-CA 和CoPc@CB 的CO2RR 的電化學活性測試,在H-cell 中進行(詳見實驗部分),所有提到的電勢都轉化為vs.RHE(無iR 補償),測試電流轉化成電流密度。
循環伏安法測試曲線(圖7a)所示,CoPc@N-CA 和CoPc@CB 都顯示出對CO2的催化活性,這與線性掃描伏安法測試的結果相一致(圖7b)。
對比CoPc@CB 的性能,CoPc@N-CA 顯示出明顯增強的活性(圖7b),在相同的電位下,CoPc@N-CA 表現出更大的電流密度,這可能與其多孔、易傳質的結構相關。
CoPc@N-CA 和CoPc@CB 的電化學性能圖如圖8 所示。
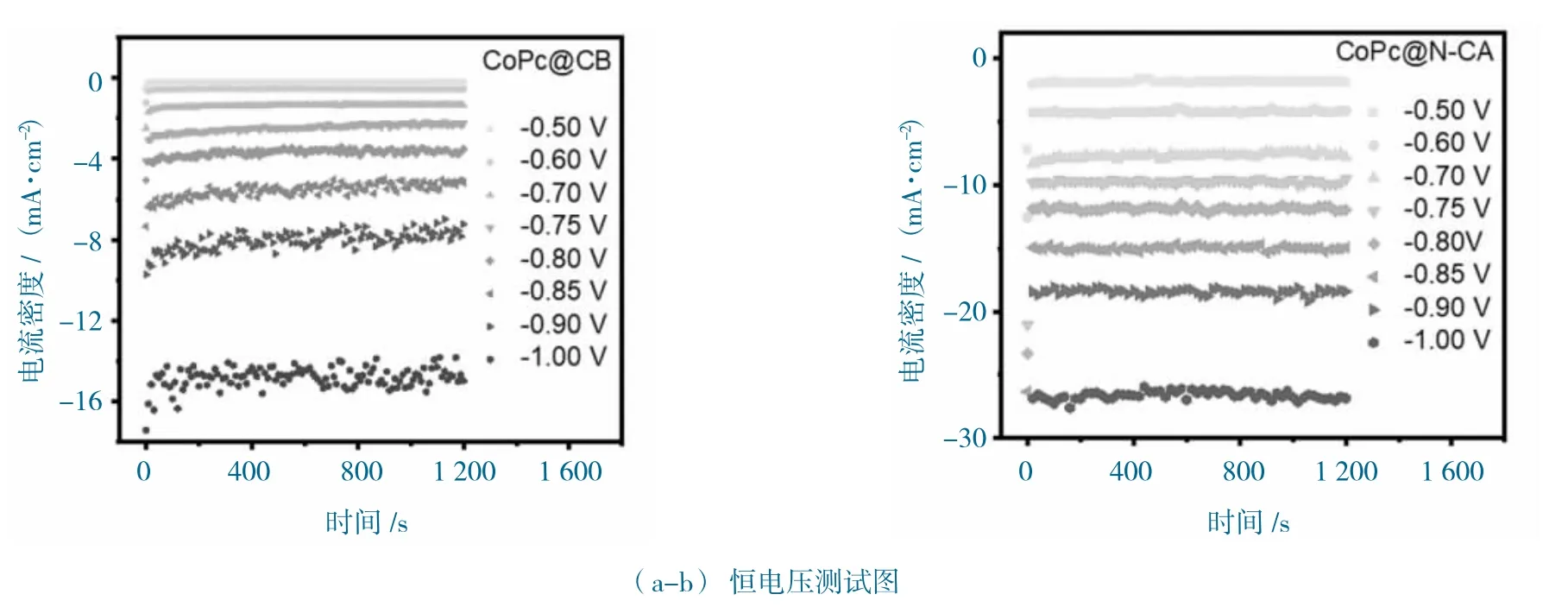
圖8 CoPc@N-CA 和CoPc@CB 的電化學性能圖Fig.8 The electrochemical performance of CoPc@N-CA and CoPc@CB
進一步恒電位測試,CoPc@CB 和CoPc@N-CA的測試曲線如圖8a-b 所示。CoPc@N-CA 在相同的測試條件下,表現出明顯增強的活性和穩定性。在-0.9 V vs.RHE 下,CoPc@N-CA 顯示-18.49 mA·cm-2的總電流,而CoPc@CB 僅顯示-7.68 mA·cm-2的總電流。在線氣相分析只發現了H2和CO 產物,1H 核磁分析未發現液相產物。不同電位下對應CO的法拉第效率(FECO),如圖8c 所示。CoPc@N-CA和CoPc@CB 的FECO 展現出“類火山”趨勢,獲得最大值為99.05%(-0.7 V vs.RHE),而CoPc@CB 獲得最大CoPc@CB 為89.41%(-0.75 V vs.RHE),這說明N-CA 特殊的結構,可以增強位點對還原產物的選擇性。進一步計算CO 的部分電流密度(jCO),CoPc@N-CA 顯示出CoPc@CB 更高的jCO,如圖8d 所示。在-0.85 V vs.RHE 電位下,CoPc@N-CA 的jCO 達到14.40 mA·cm-2,是CoPc@CB 的4 倍(3.66 mA·cm-2),這說明CoPc@N-CA 特殊的多孔、3D 無序網絡結構,有利于促進對CO 產物的活性。CoPc@N-CA 和CoPc@CB 的電化學性能圖如圖9 所示。
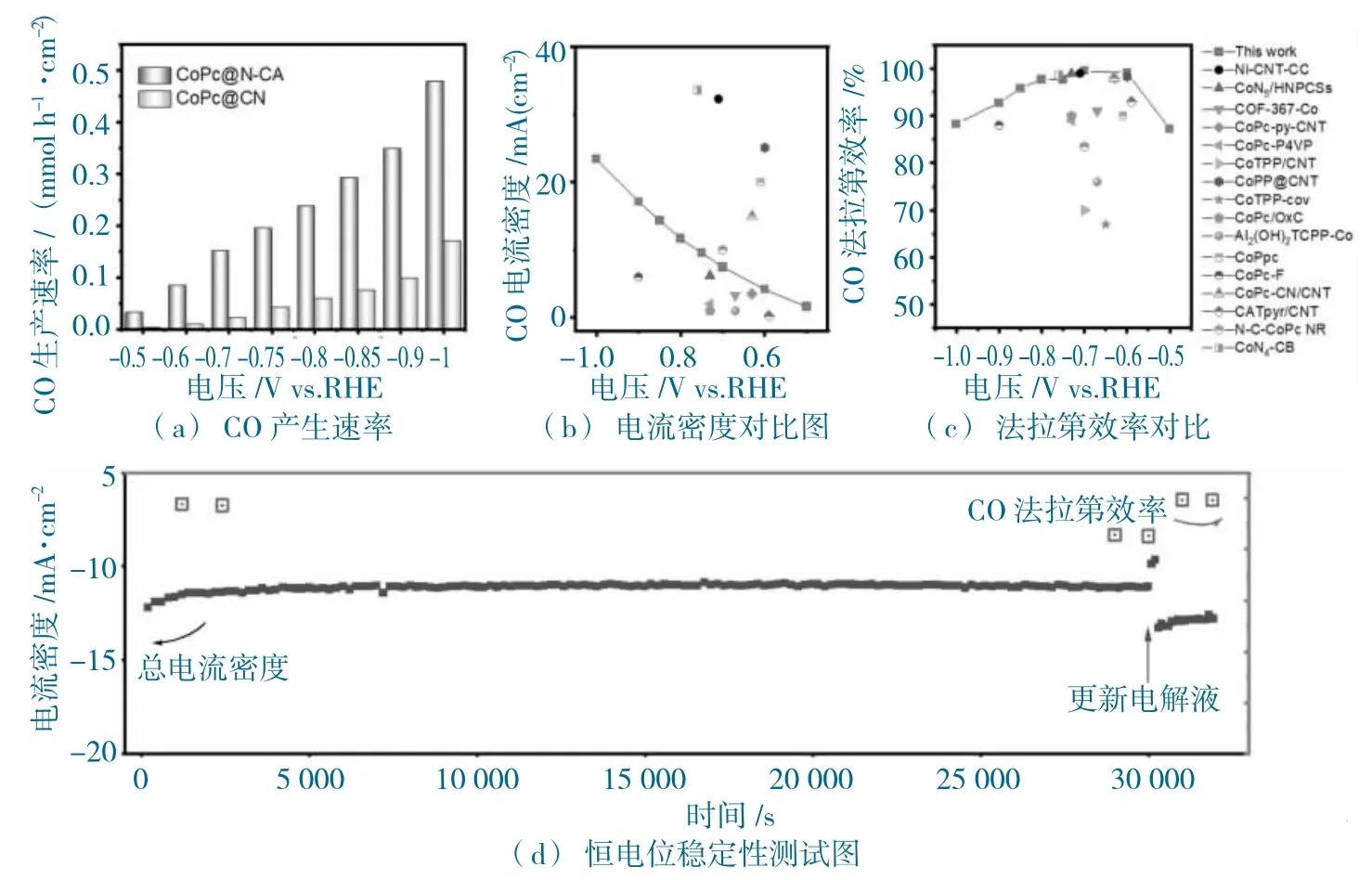
圖9 CoPc@N-CA 和CoPc@CB 的電化學性能圖Fig.9 The electrochemical performance of CoPc@N-CA and CoPc@CB
在-1.00 V vs.RHE 電位下,在CoPc@N-CA 上產CO 的速率為0.48 mmol h-1cm-2(圖9a),而CoPc@CB 僅有0.17 mmol h-1·cm-2。CoPc@N-CA 的jCO(圖9b)與FECO(圖9c),與已報道的Co 基多相分子催化劑的性能相比,明顯高于大多數催化劑的電化學性能。進一步恒電位測試發現(-0.8 V vs.RHE,圖9d),總電流穩定在大約-11 mA·cm-2,30 000 s 的測試條件下;,其CO 選擇性維持在85%以上,并且通過更新電解池,CoPc@N-CA 的電催化活性可以恢復,如圖9d 所示,突出CoPc@N-CA催化劑的高選擇性,高穩定性。
3.4 內在活性分析
樣品的活性表面積分析圖如圖10 所示。
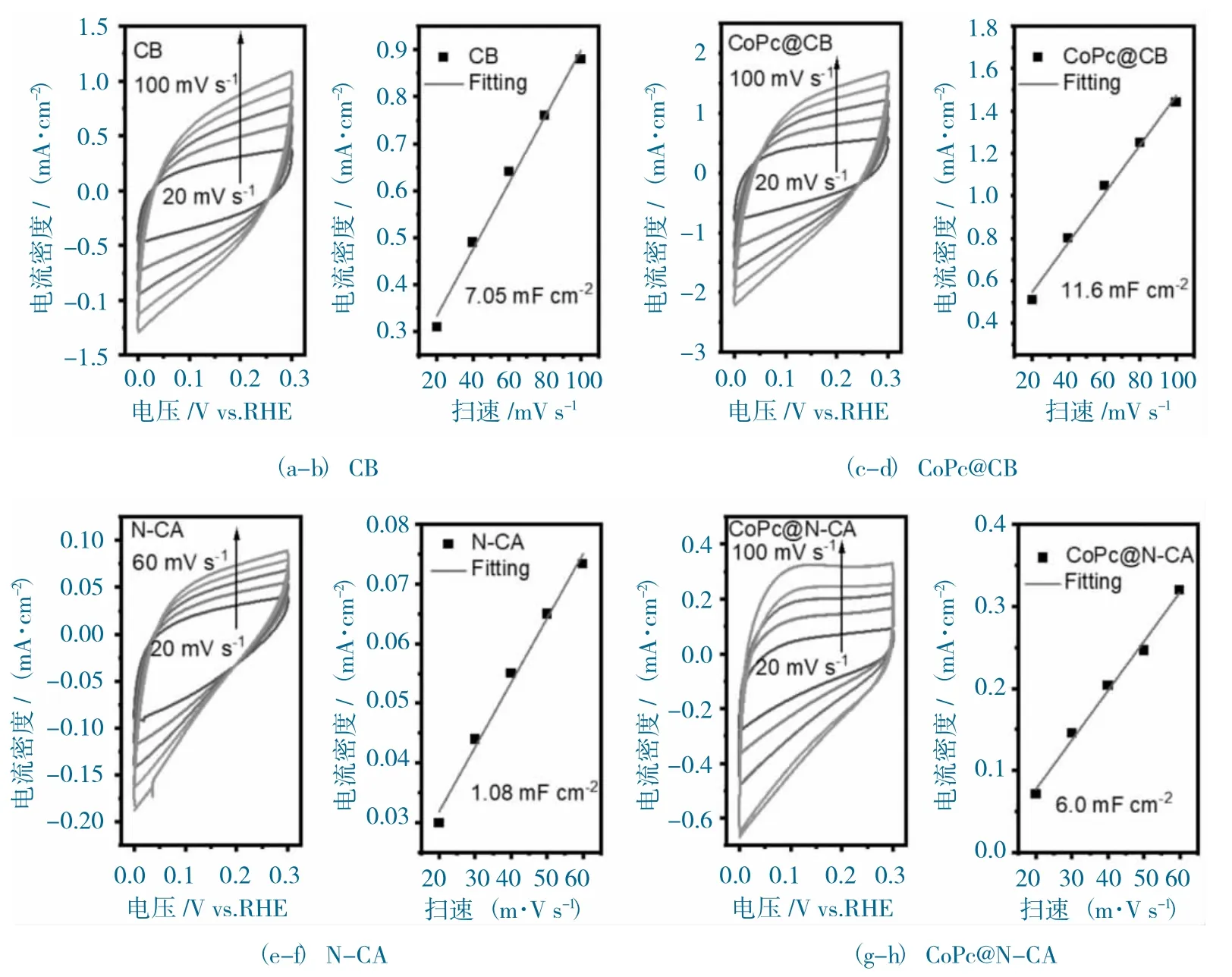
圖10 樣品的活性表面積分析圖Fig.10 The ECSA analysis of samples
測試材料的雙層電容(Cdl)值與電化學活性面積(ECSA)成正比,因此通過比較測試催化劑的Cdl 值,可以評估催化劑的活性面積。CoPc@N-CA 和CoPc@CB 的ECSA 測試在非CO2法拉第區間進行。為了探究催化劑真實的ECSA 值,首先測試了基底CB 和N-CA 的Cdl 值,分別為7.05 mF·cm-2(圖10a-b)和1.08 mF·cm-2(圖10e-f)。CoPc@N-CA 和CoPc@CB 的Cdl 值分別為11.6 mF·cm-2(圖10c-d)和6.0 mF·cm-2(圖10g-h)。扣除背景的影響,CoPc@N-CA 的Cdl 值應該為4.92 mF·cm-2,而CoPc@CB 的Cdl 值為4.55 mF·cm-2,說明CoPc@N-CA 表面暴露出更多的Co活性位點,這與N-CA 本身3D 多孔“類珊瑚狀”的結構相關。
CoPc@N-CA 和CoPc@CB 的阻抗和Tafel 斜率圖如圖11 所示。
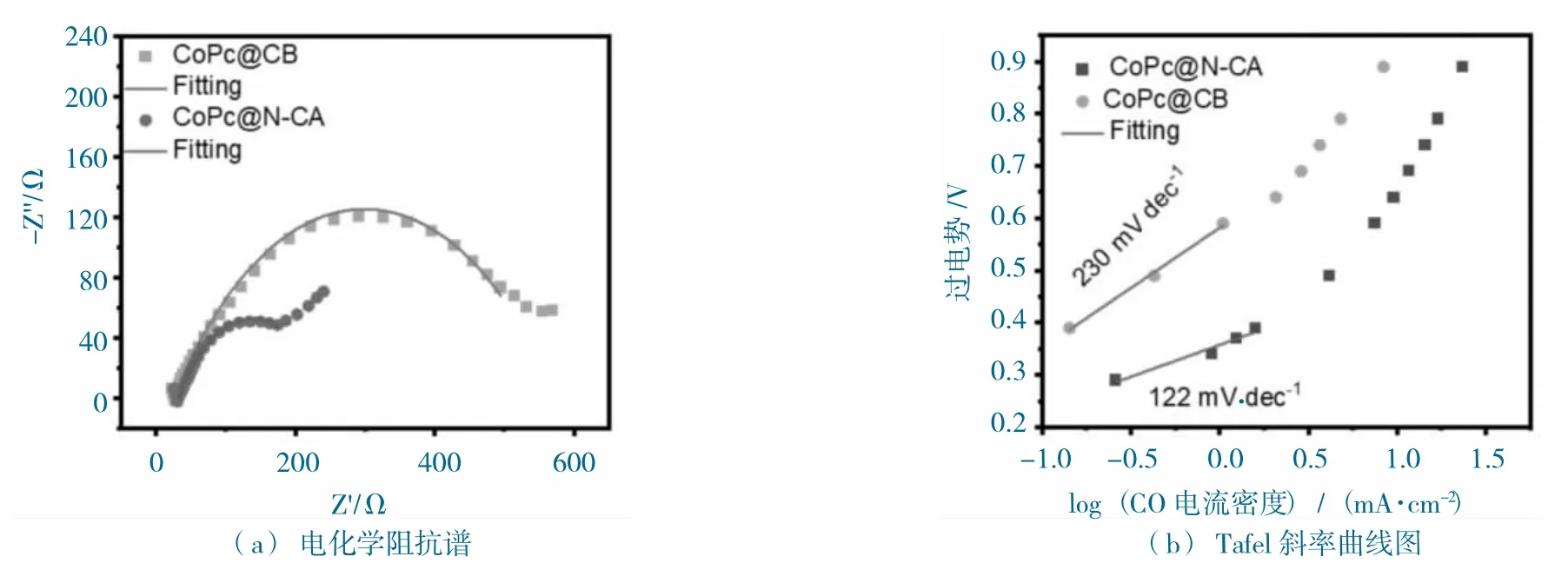
圖11 CoPc@N-CA 和CoPc@CB 的阻抗和Tafel 斜率圖Fig.11 EIS and Tafel slope diagram of CoPc@N-CA and CoPc@CB
進一步測試了催化劑的動力學性能,EIS 阻抗測試(圖11a)發現,CoPc@N-CA 擁有比CoPc@CB更小的半圓,說明CoPc@N-CA 的阻抗值比CoPc@CB 的要小,CoPc@N-CA 擁有更快的電子傳導率,這與N-CA 本身是良導體的性能相關。同時,通過催化劑的jCO,推導出CoPc@N-CA 和CoPc@CB 的Tafel 值分別為122 mV·dec-1和230 mV·dec-1(圖11b),說明CoPc@N-CA 擁有更快的CO2RR 動力學,這與基底特殊的多孔結構是分不開的。總的來說,CoPc@N-CA 擁有特殊的3D 多孔“類珊瑚狀”結構,有利于活性位點的暴露,促進CO2傳質,從而增強了CO2RR 的活性和選擇性,性能顯示優于CoPc@CB。
4 結語
通過傳統的水熱合成策略制備3D 多孔“類珊瑚狀”結構的N-CA,并通過非共價鍵固定技術,將CoPc 穩定于N-CA 的表面,形成CoPc@N-CA催化劑。基礎表征測試證明CoPc 均勻負載于N-CA 的表面,形成均一的活性位點。CoPc@N-CA催化劑中特殊的多孔結構,促進了位點的暴露和CO2的傳質,以及N-CA 本身良導體的特質加快了CO2RR 的反應動力學,因此,該催化劑表現出對CO2分子良好的電化學還原性能。在較寬的電勢窗口-0.5~-1.0 V vs.RHE 下,CoPc@N-CA 對CO 的選擇性>80%,最高為99.05%。同時,在-0.85 V vs.RHE 測試電位下,CoPc@N-CA 對CO 的部分電流密度達到14.40 mA·cm-2,約是CoPc@CB(3.66 mA·cm-2)催化劑的4 倍。本工作不僅為多孔碳氣凝膠催化劑上設計高活性的Co 位點提供了一種簡單有效的策略,同時該研發的催化劑的高催化活性和好的穩定性說明著其在大規模應用方面極具潛力。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
