礦物材料在燃料電池質子導體中的研究進展
2023-12-22 10:10:38李俊升黃鴻鑫楊園園陳佳淇
金屬礦山 2023年11期
李俊升 黃鴻鑫 楊園園 陳佳淇
(武漢理工大學化學化工與生命科學學院,湖北 武漢 430070)
從能源危機到雙碳目標的提出,無一不預示著以傳統礦石燃料為主的能源消費結構將會引起國家能源安全隱患以及社會經濟體系不可持續發展。 新型綠色能源的開發利用是促進社會經濟發展、保障國家能源安全以及保護生態環境的重要途徑。 氫氣(H2)得益于其極小的分子量而表現出高達120 MJ/kg 的質量比能量密度,同時由于氫氣具有清潔無污染、資源豐富、來源廣泛、利用形式多樣、可再生性等特性,使其成為目前最為理想的一種綠色能源。 因此,氫能的開發利用是保障國家能源安全、更快實現“碳中和”目標以及更快完成能源低碳化轉型的重要途徑。燃料電池作為氫能的主要應用載體,其通過電化學反應將H2與O2中所含的化學能轉化為電能,具有極高的能量利用率,廣泛應用在交通運輸、集中式發電站以及便攜式設備等領域。 燃料電池根據離子傳導種類可以劃分為不同的類型,如通過質子(H+)在電池內部進行傳導的質子交換膜燃料電池(PEMFC)和磷酸燃料電池(PAFC);通過氧離子(O-2)進行傳導的固體氧化物燃料電池(SOFC) 以及通過碳酸根()進行傳導的熔融碳酸鹽燃料電池(MCFC)。其中PEMFC 和SOFC 由于其全固態結構不會發生電解液泄漏具有優異的安全性能,是目前燃料電池主要的發展方向[1]。 PEMFC 具有工作溫度低、啟動速度快、能量密度高以及噪聲低等特點,在交通工具、便攜式電源、潛艇動力源等領域備受青睞[2]。 然而,PEMFC 受限于高昂的成本以及儲氫運氫技術的不完善等因素,PEMFC 的廣泛應用仍然面臨著巨大的挑戰。
質子交換膜是PEMFC 的核心組件,目前主要采用全氟磺酸樹脂類有機聚合物作為質子交換膜材料。但是昂貴的原材料、復雜的生產工藝以及較高的能源消耗,使得全氟磺酸膜的成本每平米超711 美元[3]。同時,全氟磺酸膜對使用要求較高且存在熱穩定性較差、高溫下易發生化學降解等問題,使得PEMFC 在實際應用過程中受溫度、濕度以及燃料純度等因素的限制。 為了降低質子交換膜成本,部分氟化聚合物膜、非氟聚合物膜以及復合膜被廣泛研究[1]。 但這些有機聚合物質子交換膜在穩定性、機械性能、質子傳導性能以及生產成本等方面仍然面臨諸多挑戰。而對礦物材料加工制得的無機質子導體成本低廉、合成工藝簡單,相較于傳統的有機聚合物質子交換膜有著更好的物理耐久性,更有利于電池的長期運行,在PEMFC 中有著良好的應用前景。 同時無機質子導體的開發有利于礦物材料的高附加值化,并且拓寬了礦物材料的發展空間。 然而,新型無機質子導體的開發仍然需要克服質子傳導性能以及穩定性上的挑戰。因此,本文著重概述了不同類型無機質子導體的研究現狀,為新型無機質子導體的開發提供借鑒。
1 PEMFC 概述
PEMFC 以聚合物薄膜作為電解質,因此也被稱為聚合物電解質膜燃料電池,由通用電氣公司的化學研究員GRUBB 和NIEDRACH 在20 世紀50 年代發明[4]。 隨后在20 世紀60 年代被美國國家航空航天局應用在太空任務中,作為電力供應裝置為宇宙飛船、人造衛星、探測器等設備提供電力。 隨著能源危機的出現以及社會對清潔能源的需求,PEMFC 得到快速發展,尤其是在電動汽車領域。 豐田在2020 年推出的第二代Mirai 燃料電池汽車續航里程高達850 km,電堆的功率密度達到了5.4 kW/L,相較于1995年的0.11 kW/L,25 年間功率密度提高了約50 倍。新型高性能、低成本材料的開發是加速PEMFC 發展的關鍵。 PEMFC 作為一種由多種金屬材料與非金屬材料組合而成的能量轉換裝置,需要的材料種類復雜多樣。 而我國作為礦產資源總量豐富,礦物種類最齊全的國家,擁有54 種金屬礦產以及91 種非金屬礦產,為燃料電池用新型材料的開發提供了強有力的支撐。 同時新材料的開發也有利于傳統礦產資源利用效率的提高。
1.1 PEMFC 的結構組成
PEMFC 作為不同礦物材料高值化后的集成,主要由雙極板、氣體擴散層、催化劑層以及質子交換膜組成,結構如圖1 所示。
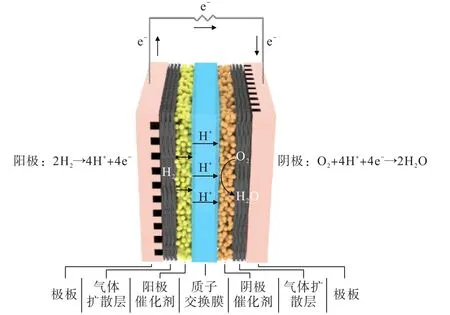
圖1 質子交換膜燃料電池工作原理及結構示意Fig.1 Working principle and structural diagram of proton exchange membrane fuel cells
雙極板是PEMFC 的關鍵功能部件之一,其表面刻有流場,因此也稱為流場板。 雙極板主要起著傳輸反應氣體、支撐電池結構、收集電流以及輸送冷卻劑等作用[5]。 為了滿足雙極板的功能,要求雙極板材料具有高導熱性、高導電性、優異的耐腐蝕性、與氣體擴散層具有良好化學和機械兼容性、良好的可加工性以及高表面平滑度等特點[6]。 根據應用領域的不同,雙極板所采用的材料也有所區別,主要有石墨雙極板、金屬雙極板以及復合雙極板三大類。 石墨雙極板是最早的一類雙極板,主要是以天然的鱗片石墨為原料,通過高溫石墨化、化學氧化、化學插層以及高溫膨脹制得膨脹石墨,然后通過對膨脹石墨進一步加工處理得到。 石墨雙極板具有優異的耐腐蝕性、良好的導熱性和導電性以及較高的化學穩定性等特點,但是易碎和多孔的特點使得石墨雙極板可加工性差、整體質量和體積較大、整體成本較高,難以滿足市場需求[7]。 以鐵礦石、鈦礦、鋁土礦為原材料,制備得到的不銹鋼、鈦合金、鋁合金等金屬雙極板具有優越的機械性能、良好的阻氣性能以及較低的生產成本等優點,是傳統石墨雙極板的有效替代品[8-10]。 然而金屬雙極板的耐腐蝕性較差,需要通過表面處理改性來提高金屬基材料的耐腐蝕性以及降低表面接觸電阻。復合雙極板將石墨和金屬材料進行復合,同時兼顧了石墨材料的耐腐性以及金屬材料的高機械性能,但復雜的制備工藝使其在大規模生產上仍然面臨巨大挑戰。
氣體擴散層作為PEMFC 核心部件之一,位于雙極板和催化層之間并將二者連接,起著促進反應氣體傳輸到電催化劑位點、改善水管理、支撐催化劑層以及收集產生的電流等作用[11]。 氣體擴散層材料的選擇需要滿足高透氣性、高導電性、高導熱性、良好的機械強度、良好的化學穩定性和熱穩定性以及耐腐蝕性等要求[12]。 為了滿足使用要求,氣體擴散層主要采用經過憎水處理的碳紙、碳布、炭黑紙或者無紡布作為多孔碳纖維基底,隨后使用導電炭黑和疏水劑在碳纖維基底上制備一層微孔層得到[13]。 此外,不銹鋼網、鈦氈、鎳網等金屬基材也可以作為PEMFC 的氣體擴散層[14-16]。
催化劑層作為PEMFC 核心部分,是進行氫氧化和氧還原的電化學反應場所。 精心設計的催化劑層需要具有優異的電子傳導能力、H+傳導能力、催化能力、耐腐蝕能力以及長的使用壽命[17]。 為了滿足上述要求,催化劑層一般由貴金屬催化劑與離聚物制備而成。 與其他貴金屬金屬相比,鉑(Pt)具有促進解離途徑反應、提高反應速率和降低吉布斯活化能的優異能力,常作為催化氫氧化和氧還原的材料[18]。 催化劑通常是先以鉑礦(自然鉑、硫化銅鎳鉑礦、硫鉑礦等)為原料,經過富集、精煉、溶解、納米化等流程制備得到3~5 nm 的Pt 納米顆粒,隨后將Pt 納米顆粒分散在高比表面積碳載體上,最后得到Pt/C 催化劑。 由于貴金屬Pt 的使用,催化劑層的成本占據了燃料電池總成本的55%[19]。 降低催化劑層的成本,可以通過優化催化劑層制備工藝和降低貴金屬 Pt 的使用量來實現。 根據涂層基底的類型和實驗步驟,兩種薄膜制備方法廣泛用于催化劑層的制備,分別是在氣體擴散層襯底上涂覆催化劑的催化劑涂層基底(CCS)工藝和在質子交換膜上涂覆催化劑的催化劑涂層膜(CCM)工藝[20]。 為了制備出更薄的催化劑層厚度、提高催化劑利用率以及建立更好的三相催化界面,催化劑主要通過CCM 工藝涂覆到質子交換膜兩側,然后與氣體擴散層通過熱壓制備得到PEMFC 核心組件“膜電極”[21]。 常用的CCM 工藝主要有干式噴涂法、轉印法、絲網印刷法、磁控濺射法等,其中轉印法是制備膜電極最為常用的方法之一。 降低貴金屬Pt 的使用量是降低催化劑成本的有效手段之一。如將Pt 與Co、Cr、Ni、Mn、Ru 等形成二元、三元甚至是四元Pt-M 合金來降低Pt 用量;以非Pt 材料為核,以Pt 或Pt-M 為殼,形成核殼結構能夠有效提高Pt 的利用率;或者將單個Pt 原子分散在載體材料上形成Pt單原子催化劑,最大程度上利用Pt 的催化能力[22-25]。
質子交換膜作為PEMFC 關鍵部件,在燃料電池運行中起著分隔燃料(H2)與氧化劑(空氣或者O2)、選擇性傳導H+并對電子絕緣、支撐催化劑層等作用。因此,質子交換膜需要具有優異的質子傳導性能、強大的阻氣能力、優異的化學穩定性和熱穩定性、良好的機械性能以及較低的生產成本等特點[26]。 目前商業化的質子交換膜主要是含氟有機聚合物,如美國DuPont 公司生產的全氟磺酸型質子交換膜,Nafion 系列膜;加拿大Ballard 公司生產的部分氟化型質子交換膜,BAM3G 膜以及美國Gore 公司生產的復合型質子交換膜,Gore-select-PTFE 增強膜等。 天然礦物材料螢石作為上游原材料,為含氟質子交換膜的生產奠定了堅實基礎。 但是由于氟化工藝復雜,含氟質子交換膜的價格居高不下,一些基于碳氫聚合物的非氟化質子交換膜如聚芳醚砜、聚苯并咪唑、聚芳醚酮、聚酰亞胺等,正在被積極研究。 這類質子交換膜具有良好的熱穩定性,環境友好且成本低,但在化學穩定性、質子傳導性能以及膜的使用壽命上還需要進一步優化改善[3]。
1.2 PEMFC 的工作原理
PEMFC 在工作時,H2經極板上的流場達到氣體擴散層,隨后通過擴散作用到達陽極側的催化劑層。在陽極催化層中,H2首先在Pt 納米顆粒表面發生解離吸附,而后吸附在Pt 表面的氫失去一個電子形成H+并脫離Pt 表面。 該過程稱為氫氧化反應,可以通過Tafel-Volmer 機制或者Heyrovsky-Volmer 機制進行,具體涉及到3 個基本反應步驟:只有吸附H2沒有電子轉移的Tafel 反應、吸附H2同時轉移一個H+和一個電子的Heyrovsky 反應以及釋放吸附氫產生H+和電子的Volmer 反應[27]。 而氫氧化形成的電子通過外電路到達陰極側,H+則穿過質子交換膜到達陰極側的催化劑層。 與此同時,O2也通過極板上的流場在氣體擴散層的作用下到達陰極側的催化劑層。在陰極催化層中,O2在Pt 的催化作用下被還原為H2O,該過程稱為氧還原反應。 實際氧還原反應歷程復雜,包含眾多反應步驟,首先是O2吸附在Pt 納米顆粒表面,然后與陽極側傳遞過來的H+和電子以直接四電子反應或者以連續二電子反應生成H2O。 在直接四電子反應中根據O—O 斷裂步驟可以分為解離機理和締合機理。 對于解離機理,O2先形成O?中間產物,O?相繼被還原成?OH 和H2O;對于締合機理,O2先被還原成?OOH,而后?OOH 被還原成H2O 和O?,O?接著被還原成?OH 和H2O。 在連續二電子反應中O2先被還原成H2O2,而后H2O2再被還原成H2O[28]。 PEMFC 在運行時涉及的反應如下所示(式中?表示催化劑中的活性位點):
陽極氫氧化反應:
Tafel 反應:
Heyrovsky 反應:
Volmer 反應:
陰極氧還原反應:
解離機理:
締合機理:
連續二電子反應:
總反應:
1.3 傳統聚合物質子交換膜所面臨的挑戰
分析PEMFC 的工作原理可以發現,H+在質子交換膜中的順利遷移是PEMFC 能夠高效運作的關鍵。質子交換膜性能的好壞決定著PEMFC 的放電功率以及使用壽命。 目前,Dupont 研發的Nafion 系列質子交換膜,如Nafion-115、Nafion-117 等占據著PEMFC 電解質膜的主要市場。 作為一種全氟磺酸樹脂類聚合物質子交換膜,Nafion 膜以聚四氟乙烯作為疏水主鏈,以含有磺酸基團的醚支鏈作為親水側鏈。 高度疏水的碳氟主鏈可以為聚合物膜提供優異的化學穩定性、良好的水穩定性以及良好的機械強度。 側鏈上親水的磺酸基團相互聚集形成彼此連通的富離子區域,為H+的快速傳導提供通道,使得Nafion 膜在80℃完全水合狀態下質子電導率可以達到0. 1 S/cm[29-30]。 然而Nafion 膜在擴大應用規模上還面臨著以下挑戰:
(1)質子交換膜的制備涉及全氟離子交換樹脂前驅體的合成以及溶液成膜工藝,整個制備工藝過程復雜、合成困難而且最終的成膜率較低,導致Nafion膜成本高昂。
(2)Nafion 膜對使用溫度和含水量要求高,最佳的使用溫度在80 ℃左右。 溫度過高會導致膜的含水量迅速下降,質子傳導性能急劇降低,同時在較高溫度下Nafion 的化學穩定性差,容易老化降解。 而較低的使用溫度一方面導致陰極氧還原反應動力學過程緩慢,需要更多的Pt/C 催化劑去促進反應進行,使得成本上升。 另一方面較低的溫度導致Pt/C 催化劑對CO、SO2等雜質氣體具有較差的耐受性,容易被雜質氣體占據活性位點,導致催化劑的活性降低甚至是失去催化活性,從而對燃料的純度有著嚴格要求,難以使用成本較低的重整氫[1]。 此外,低溫、高含水量的使用要求導致電池水管理復雜化,引起整個電池成本的上升。
(3)為了獲得更高的電池性能,目前膜電極的制備主要通過CCM 工藝將Pt/C 催化劑涂覆在Nafion膜的兩側。 然而,Nafion 膜在吸水和脫水的過程中會有10%~20%的尺寸變化,較差的尺寸穩定性容易造成催化劑從質子交換膜表面剝離,從而導致電池性能以及使用壽命變差。
因此,開發新型質子導體,進一步提高質子交換膜的性能仍然是推進PEMFC 廣泛商業化所面臨的主要任務。
2 質子在電解質中的傳導
2.1 質子傳導機制
明確質子傳導機制、了解質子傳導過程、掌握質子傳導機制判定對指導新型質子導體材料的設計至關重要。 目前質子傳導機制主要有兩種,一種是1806 年GROTTHUSS 提出的Grotthuss 機制,也稱為旋轉-跳躍機制,一種是1982 年KREUER、RABENAU和WEPPNER 提出的Vehicle 機制,也稱為運載機制[31]。 在Grotthuss 機制中H+可以圍繞氧原子快速轉動和重新定向,通過與相鄰的氧原子形成新的氫鍵而后斷裂舊的氫鍵進行傳導。 對于Vehicle 機制,H+以H2O 或NH3等分子為載體,以水合氫離子(H3O+、H5、H9)或N H+4等復合離子的形式通過擴散作用進行傳導。
2.2 質子傳導過程
對于不同性質的質子導體而言,雖然質子傳導機制都是Grotthuss 機制和Vehicle 機制,但具體質子傳導過程以及2 種傳導機制的貢獻程度存在明顯區別。在傳統全氟磺酸質子交換膜中,質子傳導機制是基于1981 年GIERKE 等人提出的離子簇—網絡模型[32]。該模型認為疏水的碳氟主鏈相互聚集形成疏水區域,親水的磺酸基團水化后相互連接形成親水性納米結構域,以反膠束離子簇形式存在。 其中離子簇的直徑為4 nm,2 個相鄰離子簇距離為5 nm,彼此通過直徑為1 nm 的通道網絡相互連接,而水和離子通過該網絡進行傳導,模式如圖2 所示。 溫度和濕度在H+傳輸過程中起著重要作用,這2 個因素決定著膜的含水量,而質子傳導機制又與膜含水量直接相關。 在全氟磺酸質子交換膜中質子在傳導過程中會同時以Vehicle 機制和Grotthuss 機制進行,但是不同含水量情況下占據主導地位的傳導機制不同,如圖3 所示[33]。在高含水量情況下Vehicle 機制占據主導地位,H+以H3O+作為載體通過被H2O 填充的通道進行整體遷移。 隨著含水量的降低,Grotthuss 機制逐漸占據主導地位,H2O 分子通過旋轉運動使得氧原子、氧原子中的空軌道與H3O+中的H+到達合適位置,而后H3O+中的H+向該H2O 分子跳躍形成新的H3O+,以此往復從而實現H+在水分子之間傳導。 當含水量進一步降低時,H+將通過離子簇表面的磺酸基團進行傳導,而由于2 個相鄰的磺酸基團距離較大,H+難以直接通過磺酸基進行傳導,需要借助位于2 個相鄰磺酸基團之間的H2O 分子以Grotthuss 機制進行傳導,這種傳導方式由EIKERLING 和KORNYSHEV 在2001 年提出,也被稱為表面機制[34]。
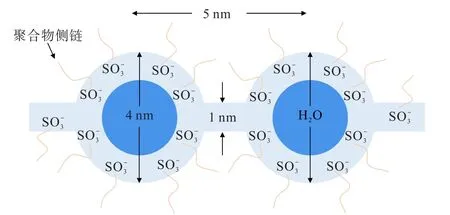
圖2 全氟磺酸質子交換膜的離子簇—網絡模型[32]Fig.2 Ion cluster network model of perfluorosulfonic acid proton exchange membrane[32]
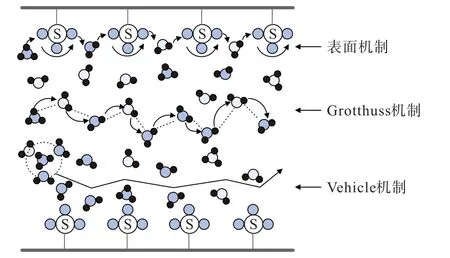
圖3 全氟磺酸質子交換膜不同質子傳導機制示意[33]Fig.3 Schematic diagram of different proton conduction mechanisms in perfluorosulfonic acid proton exchange membranes[33]
對于以礦物材料制備得到的無機質子導體,通常會存在體積內質子傳導和表面質子傳導。 體積內的質子傳導主要依賴于氧空位的存在。 質子以Vehicle機制進行傳導時,H+隨著氧離子一起沿著氧空位進行遷移;質子以Grotthuss 機制進行傳導時,H+首先圍繞著氧原子進行旋轉定位然后跳躍到相鄰的氧原子上,重復上述行為實現H+的傳導,2 種傳導機制如圖4 所示[35]。 而表面質子傳導主要依賴于表面H2O 分子的吸附。 H2O 分子會與裸露的導體表面的末端金屬陽離子結合形成化學吸附層,被吸附的水分子可能進一步解離,形成端羥基和多配位羥基,如圖5 所示[36]。 當相對濕度從0 到30%,H2O 分子與導體表面的端羥基形成氫鍵,而后繼續增長到三層,由于化學吸附層和第一物理吸附層中的強成鍵,它們在結構上是類似于冰的,因此前三層水分子稱為“類冰態水”。 當相對濕度從30%增加到60%時,冰狀結構繼續增長至飽和,其中可能存在多聚物鏈水層,形成“冰”和“液體”結構之間的過渡區。 進一步將相對濕度提高到60%以上,吸附層厚度開始呈指數增加,最外層吸附的水分子結合更加松散,呈液態水結構[37-39]。 無機質子導體表面呈現出液態水時,H+能夠以Vehicle 機制進行,當表面H2O 分子以氫鍵網絡相互連接時,Grotthuss 機制成為H+傳導方式的首選。
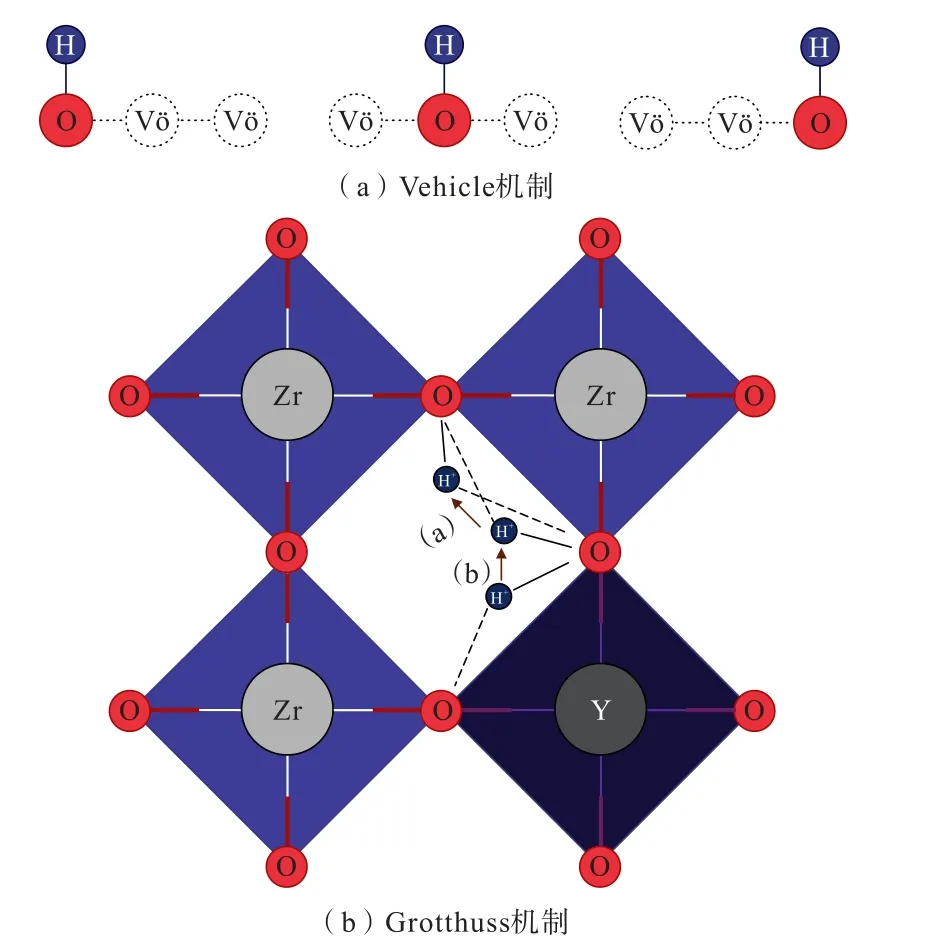
圖4 體積內質子傳導機制示意[35]Fig.4 Schematic diagram of proton conduction mechanism within the volume[35]
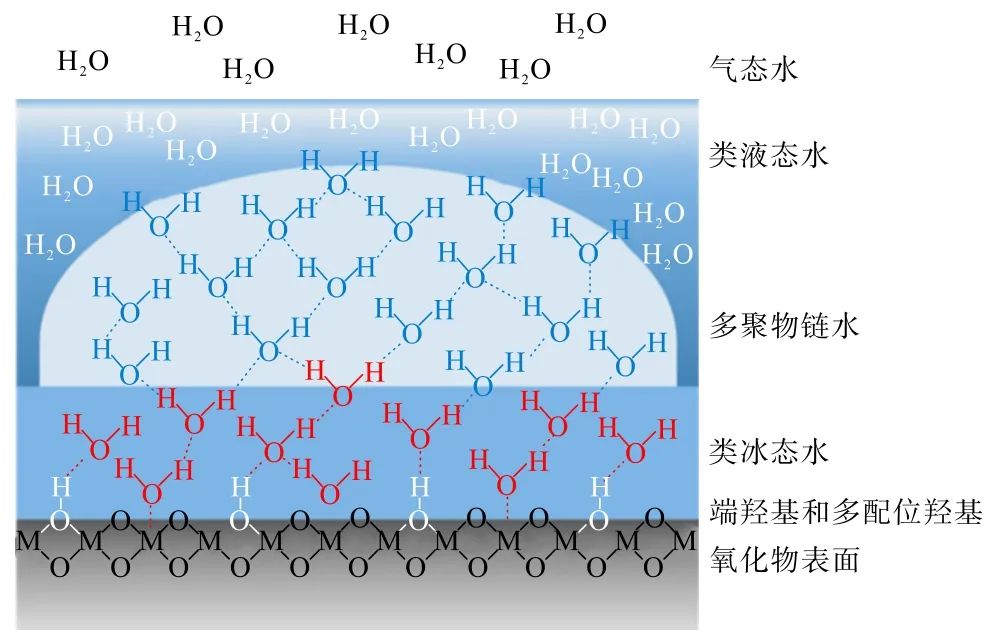
圖5 無機質子導體表面吸附水層示意[36]Fig.5 Schematic diagram of inorganic proton conductor surface adsorbed water layer[36]
2.3 質子傳導機制的判定
傳導機制的確定主要可以通過2 種方式,一種是通過質子傳導活化能的大小進行區分。 活化能可以通過測定不同溫度下質子導體的電導率而后利用Arrhenius 方程進行擬合得到。 通過公式(16)求得的活化能稱為表觀活化能,主要反映了質子傳輸時所克服的自由能勢壘。 當活化能在0.1~0.4 eV 時,Grotthuss 機制占據主導地位;活化能在0.5~0.9 eV 時,Vehicle 機制占據主導地位;活化能在0. 4~0. 5 eV時,通常考慮為2 種機制并行[40]。
式中,σ是質子電導率,S/cm;T是絕對溫度,K;A是指前因子;Ea是活化能,eV;k是玻爾茲曼常數,通常為1.38×10-23J/K。
另一種則是通過利用同位素效應對比H 傳導和H 同位素D 傳導的區別。 H/D 以Grotthuss 機制進行傳導時,O—H/O—D 鍵斷裂,H/D 躍遷到相鄰的氧上,形成新的O—H/O—D 鍵。 在經典理論模型中,質子跳躍速率跟O—H/O—D 的伸縮頻率成正比,而伸縮頻率又與根號質量分之一成正比,且活化能Ea與同位素無關,由此可得σH/σD理論值為1. 4[41]。一般來說,同位素效應數值的大小可以反映質子的傳導機制,當σH/σD數值在1.4 及以上時,質子以Grotthuss 機制進行傳導,σH/σD數值在1 左右時,質子以Vehicle 機制進行傳導。 但是在實際過程中由于同位素質量不同,H/D 物種在其所在勢阱中的零點能E0不同,因此H 和D 傳導的活化能不同,從而σH/σD的數值會出現偏離理論值1.4 較多的情況。
3 礦物材料在質子導體中的應用
PEMFC 的發展在一定程度上受限于質子交換膜的發展。 目前,廣泛應用在PEMFC 中的有機聚合物質子交換膜存在成本高昂、生產工藝復雜且熱穩定性差等問題。 而無機質子導體具有優良的穩定性以及可調控的質子傳導行為,被認為是理想的PEMFC 電解質材料。 無機質子導體的設計合成離不開礦物材料的支持,在目前已經開發的無機質子導體中主要有氧化物、固體酸、金屬鹽三大類,涉及到眾多種類的礦物材料,其中稀土礦、鋇礦、鋯礦以及磷礦是無機質子導體的主體材料來源。 同時一種無機質子導體會涉及多種元素,與多種礦物材料相關,本文以其共同涉及的某一元素進行分類,將目前的無機質子導體分為稀土礦衍生的質子導體、鋇礦衍生的質子導體、鋯礦衍生的質子導體以及磷礦衍生的質子導體四大類。各種質子導體材料的性能見表1。
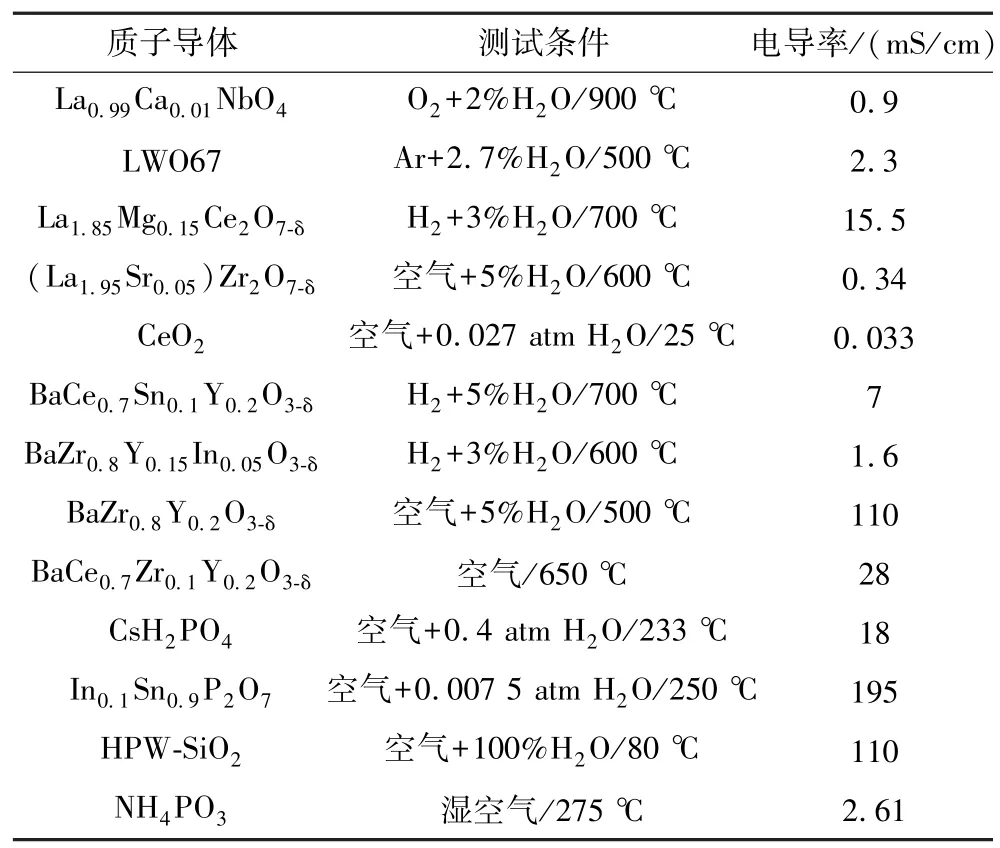
表1 各種質子導體材料不同測試條件下的質子電導率Table 1 Proton conductivity of various proton conductor materials under different test conditions
3.1 稀土礦衍生的質子導體
稀土礦是一種戰略資源,目前氟碳鈰礦、獨居石礦、磷釔礦以及風化殼淋積型礦是工業提取稀土元素的主要礦物,稀土元素具有優異的光、電、磁等物理特性,在航空航天、石油化工、新能源、農業、紡織等領域具有不可替代的地位。 稀土元素活潑的化學性質使其能夠生成極穩定的氧化物,該氧化物具有優異的化學穩定性和熱穩定性,能夠在高溫和極端環境下保持結構的穩定性,這種穩定性對于電解質材料來說至關重要。 在17 種稀土元素中,鑭的氧化物和鈰的氧化物可以作為電解質材料用來傳導H+或者O2-,而釔、鈧、釤、銪、釓、鐿等稀土元素常作為電解質的摻雜劑,用于提高電解質的整體性能。
3.1.1 鑭基質子導體
在氧化物型質子導體中質子的傳導主要依賴H2O 分子與氧空位結合形成質子缺陷,或者H2還原晶格氧形成質子缺陷。 因此氧化物型質子導體需要本身具有晶格氧缺陷,或者能夠通過低價元素摻雜產生氧空位。 螢石結構和燒綠石結構具有開放式結構,能夠接受低價元素的摻雜而保持結構穩定性,是較為理想的氧化物晶型。 鑭基質子導體主要有兩大類一種是基于螢石結構的鈮酸鑭(LaNbO4)、鎢酸鑭(La6WO12)、鈰酸鑭(La2Ce2O7),還一類是基于燒綠石結構的鋯酸鑭(La2Zr2O7)。
3.1.1.1 鈮酸鑭
鈮酸鑭(LaNbO4)作為一種質子導體,其在490℃到520 ℃之間會發生相變,從單斜相轉變為四方相[42]。 這2 種晶體結構的LaNbO4均表現出質子傳導性能,但是相比于四方晶型,質子在單斜晶型中具有更高的各向異性,表現出更高的質子傳導活化能[43]。 2006 年HAUGSRUD 等人研究LaNbO4和摻雜LaNbO4的質子傳導性能,純的LaNbO4在950 ℃時電導率約為0.3 mS/cm,而性能最好的1.0 % Ca摻雜的LaNbO4在900 ℃下電導率也僅約為0. 9 mS/cm[44]。 因此,LaNbO4材料的研究主要聚焦于如何進一步提高其質子傳導性能。 目前主要的策略就是通過摻雜取代部分La、Nb 或者兩者同時部分取代,以增加氧空位濃度的形式來提高其質子電導率。Ca、Sr、Zn、Mg、Ba、Gd 等元素常用來取代La 位點,Ga、Ge、Si、B、Ti、Zr、P、Al、Sn 等元素常用取代Nb 位點,然而這些摻雜的LaNbO4并沒有觀察到明顯增強的質子傳導性能,甚至有的質子電導率反而下降[45-49]。 這是由于摻雜元素溶解度的限制,La 位點上摻雜元素的溶解度僅為0.5%~2%,Nb 位點上的摻雜元素溶解度最高為3%,如此低的摻雜濃度使得氧空位濃度的提高并不理想。 因此提出新的改性策略是提高LaNbO4質子傳導性能的關鍵。
3.1.1.2 鎢酸鑭
鎢酸鑭(LWO)是一種混合導體,同時具有質子傳導性能和電子電導性能。 LWO 電導率特性的研究首次報道是在2001 年, SHIMURA 等人研究了La5.8WO11.7的電導率特性,發現其在濕潤H2氛圍下表現出混合質子電子電導,在800 ℃下總電導率高達50 mS/cm[50]。 2007 年,HAUGSRUD 通過交流阻抗研究了LWO 在濕H2和濕D2氛圍下的電導率,在800 ℃以下時,濕H2下的電導率高于濕D2下的電導率,表現出明顯的同位素效應,說明當溫度低于800℃且在潮濕氛圍下LWO 的電導率主要受質子傳導控制[51]。 研究表明未摻雜LWO 在800 ℃潮濕氛圍下質子電導率為5 mS/cm,而在干燥氛圍下LWO 表現出一定的氧離子傳導,在高溫下LWO 以電子導電為主,高溫氧化氛圍下以p 型空穴傳導,高溫還原氛圍下以n 型電子傳導[52-53]。
為了提高LWO 的質子傳導性能,傳統的摻雜方式被廣泛研究。 然而無論是使用Ce、Ca、Sr 等元素對La 位點進行摻雜,還是Mo、Nb、Cr 等元素對W 位點進行摻雜,均未顯著提升LWO 質子傳導性能[54-57]。同時W 位點的摻入還會導致LWO 材料電子傳導性能的提高。 通過增加La/W 比來增加LWO 氧空位濃度,從而增加其質子傳導性能是一種有效手段。 在LWO 晶體結構中,當La/W 小于7.0 時,W 起施主摻雜作用部分La3+位點被W6+占據,W 原子取代La 位點提高了LWO 的結構穩定性,但是電荷中和降低了晶體結構中的本征氧空位濃度。 而LWO 中的La/W可以通過合成過程進行控制。 在此之前,MAGRASó等人通過冷凍干燥的方法合成了La/W 5.3~5.7 的單相LWO 樣品,并對其晶相和質子電導率進行了研究。 研究發現LWO 的質子傳導性能隨著La/W 的增加而增加,但是當La/W 小于5.3 時觀察到La6W2O15的形成,而當La/W 大于5.7 時觀察到了La2O3的形成[58]。 在2017 年之前由于制備工藝的原因,La/W最大被限制在了5.7。 2017 年KOJO 等人通過1 700℃的高溫燒結獲得了La/W 在6.3~6.7 范圍內的單相LWO,當La/W 在6.7 時LWO 性能最佳,CO2氛圍下具有良好的化學穩定性,在500 ℃時質子電導率達到了2.3 mS/cm,同時700 ℃以下時電子電導和空穴電導被充分抑制[59]。 上述研究結果表明LWO 有望通過改進制備方法來提高其應用價值。
3.1.1.3 鈰酸鑭
鈰酸鑭(La2Ce2O7)可以認為是50%的La2O3摻雜的CeO2,因此在成相過程中會發生缺陷反應,使得La2Ce2O7本身就含有氧空位,從而能夠在不摻雜的情況下傳導質子。 SUN 等人研究表明純La2Ce2O7在濕空氣(3% H2O)550 ℃下質子電導率可以達到66.8 μS/cm,同時發現總電導率隨水蒸氣分壓的增加而顯著增加[60]。 由于Ce4+在還原氛圍下會被還原成Ce3+從而產生電子電導,因此La2Ce2O7是混合質子電子導體。 而且隨著溫度的升高La2Ce2O7中電子電導占據主導地位,在2.5% H2O 濕空氣800 ℃時總電導率便高達14.1 mS/cm[61]。 而純La2Ce2O7質子電導率偏低,難以應用,研究表明在La 位點上摻雜Li、Na、K、Rb、Cs 等堿金屬能夠顯著提高La2Ce2O7的質子傳導性能,且電導率隨摻雜堿元素離子半徑的增大而增大,而隨銫摻雜而減小,La1.85Rb0.15Ce2O7-δ性能最佳,在800 ℃的濕H2中表現出6.21 mS/cm 的電導率,且以La1.85Rb0.15Ce2O7-δ基的單電池,在700℃下表現出高達1 031 mW/cm2的峰值功率,穩定放電時間超過200 h[62]。 當使用堿土金屬Mg 對La2Ce2O7進行摻雜時,不僅可以增加La2Ce2O7電導率,還能作為燒結助劑降低La2Ce2O7燒結溫度。 WU等人在1 300 ℃燒結5 h 的條件下成功制備出全致密的La1.85Mg0.15Ce2O7-δ電解質膜[63]。 此外,在700℃的濕H2(3% H2O)中La1.85Mg0.15Ce2O7-δ的電導率最高為15.5 mS/cm,并且對CO2和H2O 保持優異的化學穩定性,同時基于La1.85Mg0.15Ce2O7-δ的燃料電池在700 ℃時表現出高達897 mW/cm2的峰值功率密度[63]。 對La2Ce2O7的La 位點進行摻雜是提高La2Ce2O7整體性能至關重要的手段,而進一步降低La2Ce2O7使用溫度是提高其應用價值的必經之路。
3.1.1.4 鋯酸鑭
早在1997 年LABRINCHA 等人就報道了純相的La2Zr2O7具有質子傳導性能,但是其質子導電性較差,在900 ℃下僅有4.2 μS/cm[64]。 TAKAHISA 等人研究發現純相的La2Zr2O7不能溶解H2O 蒸氣,從而導致質子傳導性能較差,而使用Ca2+摻雜的La2Zr2O7具有較好的H2O 蒸氣溶解能力[65]。 電動勢測試結果表明, 在 200 ~ 800 ℃ 范圍, ( La1.98Ca0.02)(Zr1.98Ca0.02)O7-δ是純離子導體,而且在600 ℃以下是純質子導體,在600 ℃以上開始出現O2-傳導,但是O2-傳導較弱,在800 ℃時O2-的轉移數也僅為0.18,質子傳導占據主導地位。 通過對La 位點和Zr位點使用堿土金屬進行摻雜取代,能夠顯著增加La2Zr2O7的質子傳導性能。 TAKAHISA 等人的研究表明,由于La2Zr2O7燒綠石相的特殊結構,導致僅被La 原子包圍氧原子顯示出高堿度,因此La 位點摻雜的(La2-xMx)Zr2O7-δ中質子溶解度要比Zr 位點摻雜的La2(Zr2-xMx)O7-δ(M=Mg、Ca、Sr、Ba)高出3 倍[66]。同時摻雜堿土金屬的種類對La2Zr2O7的質子傳導性能也有所影響,BJ?RKETUN 等人研究發現Ca 和Sr與質子的相互作用較弱,對質子的傳導起促進作用,而Mg 和Ba 的存在對質子的遷移有著負面影響[67]。此外,晶粒尺寸對La2Zr2O7基材料的電導率也有一定影響,在(La1.95Sr0.05)Zr2O7-δ中總電導率隨著晶粒尺寸的增加而單調增加[68]。 總體而言,燒綠石結構的La2Zr2O7質子傳導性相較于螢石結構的La 基材料偏低,但是其具有良好的燒結特性[69]。
3.1.2 鈰基質子導體
鈰基質子導體主要有三類,分別是上文介紹過的螢石結構La2Ce2O7、鈣鈦礦結構的鈰酸鋇(BaCeO3)以及氧化鈰(CeO2)。 BaCeO3將會在下一節進行詳細介紹,本節主要是介紹CeO2在質子導體中的研究進展。 CeO2具有螢石結構,因其優越的儲存和釋放O2的能力而被廣泛用作催化領域的催化劑、載體或促進劑。 此外CeO2還是SOFC 中一種主要用來傳導O2-的電解質。
將材料納米化時,由于擴散長度短以及界面密度高而表現出顯著的尺寸效應。 作為高溫氧離子導體的CeO2通常情況下認為是不良質子導體,但是在濕潤氛圍中CeO2的納米晶在中低溫下表現出可觀的質子傳導性。 2010 年,AVILA-PAREDES 等人通過放電等離子燒結技術制備了平均晶粒尺寸約為15 nm的Ce1-xGdxO2-δ(x=0.5、10 和20%)納米晶陶瓷,研究了其在30~200 ℃溫度范圍內質子傳導與Gd 摻雜水平之間的關系[70]。 研究結果表明,在低于200 ℃的溫度下,電導率與Gd 摻雜水平無關,當溫度高于200℃后,離子電導率隨著Gd 摻入量的增加而增大,這得益于較高的Gd 摻雜水平引起的較大濃度的氧空位。 基于上述結果,AVILA-PAREDES 等人認為在納米晶螢石結構材料中質子的傳導與體相缺陷無關,質子可能沿著晶界進行傳導。 然而,2011 年SHIRPOUR 等人研究了晶粒尺寸在40 nm 的納米晶和晶粒尺寸在300~500 nm 的微晶的導電性能[71]。 研究發現微晶樣品的電導率在潮濕和干燥氛圍下沒有變化,而納米晶樣品在潮濕氛圍下表現出明顯比干燥氛圍下更高的電導率,熱重分析進一步表明納米晶CeO2在200 ℃以下表現出明顯的吸水性,結果表明晶界和開放性空隙均有可能對質子的傳導起積極促進作用。隨后,GREGORI 等人通過脈沖激光沉積(PLD)生長制備了全致密外延納米晶CeO2薄膜,以及利用旋涂法制備了多孔納米晶CeO2薄膜,探討了孔隙率對表面質子傳導的影響。 研究結果顯示全致密外延納米晶CeO2薄膜的電導率在干濕條件下差異不大,但多孔納米晶薄膜的電導率在暴露于300 ℃以下的潮濕大氣中時顯著增加[72]。 這些結果表明,開放性空隙或者裂縫是影響質子在中低溫下通過金屬氧化物傳輸的核心因素,而不是大的晶界密度。
2018 年,MANABE 等人利用電化學阻抗譜研究了潮濕條件下多孔CeO2的表面傳輸特性。 對于相對密度為60%的低致密度樣品,通過等效并聯電路可以很好地提取晶粒和晶界的內部和表面電導率[73]。 研究結果顯示在潮濕條件下的表面質子電導率隨著溫度的降低而增加,表明吸附水在CeO2表面的質子傳輸中起著重要作用。 400 ℃時顆粒體表面(內部)的電導率對水分壓的依賴性表明,促進表面質子傳輸的是水分子而不是離解的水。 上述這些工作清楚地表明,吸附水在CeO2的表面質子傳導中起著重要作用,特別是在低溫下。 最近,SIMONS 等人利用噴涂法制備了CeO2薄膜,通過對比退火前后CeO2薄膜的電導率發現,退火后的薄膜具有更高的電導率[74]。 他們認為薄膜退火后增加了晶態CeO2的比例以及晶粒尺寸,從而增加了高導電路徑,表明晶界實際上是有助于質子的傳導。 同時,作者證明由于CeO2緩慢的水合動力學過程,室溫下潮濕大氣中獲得穩定電導率所需的時間長達76 h,這意味著動力學可能在之前的研究中抑制了質子傳導,從而解釋了迄今為止報告中CeO2電導率的強烈波動。
3.2 鋇礦衍生的質子導體
鋇的化學性質相當活潑,能與大多數非金屬反應,在自然界中難以以單質形式存在,主要以礦物形式存在。 鋇礦主要成分是鋇的鹽類,主要產出于巖漿巖、變質巖中。 鋇礦可以分為重晶石(BaSO4)、毒重石(BaCO3)和天青石(BaSO4)3 種。 我國的鋇礦資源豐富,擁有全球29%重晶石的儲量,鋇礦儲量和產量目前均居世界首位,因此鋇礦的開發利用對我國礦物產業的發展起著重要作用。 而鋇基材料的用途十分廣泛,玻璃、陶瓷、涂料、醫藥、新能源等行業均離不開鋇基材料。 鋇礦衍生的質子導體主要是鈣鈦礦材料,鈣鈦礦最初是指在1839 年發現的鈦酸鈣(CaTiO3)礦物,后來泛指與CaTiO3具有相同結構的材料。 理想的鈣鈦礦通式為ABO3,結構可以認為是面心立方晶格,角原子為A 原子,面上為O 原子,B 原子最終確定了位于晶格中心的結構。 其中A 位點為半徑較大的低價態金屬陽離子,如一價的Na+、K+,二價的Ca2+、Sr2+、Ba2+,三價的Fe3+、Gd3+、La3+;B 位點為半徑較小的高價態金屬陽離子,如W5+、Nb5+、Ce4+、Zr4+、Co3+等[75]。 A、B 位點不同金屬離子組合得到的鈣鈦礦材料表現不同的絕緣性、光學性能、鐵電性、催化性能以及離子傳導性[76]。 從1981 年IWAHARA等人發現SrCe1-xMxO3-δ(M=Yb、Y、Sc)在600~1 000℃的H2或濕空氣氛圍下表現出大于10-4S/cm 的質子傳導性能開始,具有質子傳導性能的鈣鈦礦得到了廣泛研究,其中BaCeO3以及鋯酸鋇(BaZrO3)因其良好的化學穩定性、優異的質子導電性和簡單的制備工藝而備受關注[77]。
3.2.1 鈰酸鋇基質子導體
BaCeO3是鈣鈦礦結構,Ba 在A 位點,Ce 在B 位點,具有較高的質子電導率,在中溫下比鈰酸鍶(Sr-CeO3)的質子電導率高出一個數量級[78]。 這是由于影響質子導電性的化學和結構參數決定的。 未摻雜的BaCeO3具有Pnma空間群,在高溫下表現出理想的立方結構。 低溫BaCeO3中存在的2 個結晶學上不同的氧位點在高溫下變得能量等價[79]。 一般而言,能量不相等的氧位點會使得水化行為更加復雜,而BaCeO3在高溫下克服了這一點,從而導致電導率的增加。 盡管SrCeO3與BaCeO3具有相同構型,但與BaCeO3相比,SrCeO3的晶體結構表現出較大的立方對稱性扭曲,從而在加熱到1 000 ℃時仍保持正交結構,存在2 個能量不相等的氧位點,導致導電性下降[80]。
優秀的電解質材料應滿足3 個要求,分別是高離子電導率、強化學穩定性和良好的燒結性。 BaCeO3基質子導體普遍擁有較高的離子電導率(在600 ℃時電導率≥0.01 S/cm),而且具有良好的燒結性,但是由于BaCeO3具有天然的堿性,很容易與酸性氣體如CO2、 SO2以及水反應生成BaCO3、 BaSO4和Ba(OH)2,阻礙質子的遷移,導致材料發生較大的熱膨脹,從而降低燃料電池的性能[81]。 BaCeO3在較低溫度下的分解,嚴重阻礙了其在實際中的應用,比如在1 041 ℃以下與CO2發生反應,在403 ℃以下與水蒸氣發生反應[82]。 以20% Gd 摻雜的BaCeO3(BCG20)為電解質組裝而成的燃料電池在600 和700 ℃、含50%水蒸氣環境中可以穩定運行1 000 h[83]。 但是BHIDE 和VIRKAR 的研究結果顯示,在400 ℃以下,以BCG20 為電解質的燃料電池僅運行168 h,BCG20 就會與水蒸氣發生反應而分解[84]。
MATSUMOTO 等人將不同的三價稀土離子摻入BaCeO3中,用以改變BaCeO3的性能。 研究發現摻雜BaCeO3的電導率和化學穩定性由摻雜離子半徑決定,摻雜離子半徑越大,離子電導率越高,但化學穩定性降低[82]。 因此,摻Y3+的BaCeO3表現出最佳的電導率性能,摻Sc3+的BaCeO3表現出最佳的化學穩定性,然而Sc 昂貴的價格阻礙了其廣泛應用。 對于化學穩定性的增強,很難找到比Sc3+半徑更小的三價稀土陽離子,而Al3+的半徑僅為54 pm,遠小于Sc3+的半徑(75 pm),且Al 來源廣泛價格低廉。 SHIN 等人的研究表明Al3+摻雜的BaCeO3表現出良好的化學穩定性,且當Al3+摻雜量超過20%時Al3+發生溶出,但其在400~470 ℃的電導率接近Y 摻雜的BaCeO3[85]。對Ce 位點進行雙金屬摻雜, 在BaCe0.8Y0.2O3-δ(BCY20)的基礎上引入具有更高電負性的金屬元素,能夠有效降低BCY20 的堿性,從而使BaCeO3基材料對CO2和H2O 表現出更高的化學穩定性。 BI等人將Ta 和Y 共摻雜到BaCeO3的Ce 位點中形成BaCe0.7Ta0.1Y0.2O3-δ,該材料在沸水中煮6 h 能夠保持結構的穩定性,且700 ℃時在100% CO2氛圍下表現出足夠的化學穩定性,同時以BaCe0.7Ta0.1Y0.2O3-δ作為電解質組裝的燃料電池在700 ℃的功率密度可達142 mW/cm2[86]。 XIE 等人利用固相反應法制備了Sn、Y 共摻雜的BaCe0.7Sn0.1Y0.2O3-δ,對H2O 和CO2均有很好的化學穩定性,在700 ℃濕H2氛圍下電導率可以達到7 mS/cm。 同時由于Sn 的摻入提高了電解質的可燒結性,相比于BCY20 在1 400 ℃燒結后僅有83.3%的相對密度,BaCe0.7Sn0.1Y0.2O3-δ燒結后的相對密度可以達到 96. 0%。 而且以BaCe0.7Sn0.1Y0.2O3-δ為電解質的燃料電池在700 ℃功率密度達到了470 mW/cm2,界面電阻也僅為0. 13 Ωcm2,表明該電解質材料與電極之間具有良好的相容性[87]。 除了采用金屬離子進行摻雜改性外,還可以通過在BCY20 電解質表面脈沖激光沉積一層BaZr0.8Y0.2O3-δ(BZY20),在保護BCY20 的同時不會顯著影響其導電性[88]。 尋找合適的途徑來提高BaCeO3基電解質的化學穩定性,同時保持較高的質子電導率,是實現質子傳導燃料電池在低溫下運行的關鍵。
3.2.2 鋯酸鋇基質子導體
BaZrO3具有優異的物理性能和高導熱性能,與BaCeO3相比具有更低的熱膨脹值、更好的穩定性以及更高的機械強度,能夠在CO2和H2O 氛圍下穩定存在[89]。 但是較差的燒結性能使BaZrO3作為電解質面臨著低相對密度以及高晶界電阻等問題。 而較高的燒結溫度或者較長的熱處理時間會導致BaO 的蒸發,從而引起A 位點占有率的降低,導致氧空位的濃度下降,從而對整體的導電性性能產生不利影響[90]。 因此,BaZrO3基電解質的研究方向主要是提高BaZrO3基的電導率以及燒結性能。 為了提高Ba-ZrO3基電解質的電導率,一般采用低價元素進行摻雜來提高氧空位濃度。 由于三價稀土元素Y3+的摻入不會改變質子缺陷的水化熱和遷移率,因此Y3+被認為是BaZrO3中B 位點最好的摻雜劑。 20% Y3+摻雜的BaZrO3具有較高的化學穩定性和體相質子電導率,當Y3+摻雜量超過20%時將會導致激活能增加從而引起電導率下降,因此BZY20 是目前BaZrO3基材料的主要研究對象[80]。
為了獲得高性能BaZrO3電解質,一般從使用燒結助劑、金屬離子共摻雜以及探索新的制備工藝3 個方面提高BaZrO3的燒結性能。 在BZY20 中添加1%的ZnO 作為燒結助劑,焙燒時Zn 進入晶格與BZY20形成固溶體,從而使得BZY20 的燒結溫度從1 700 ℃降低至1 325 ℃,致密度達到96%,在600 ℃以上時,總電導率可以達到1 mS/cm,明顯高于未加ZnO 的BZY20[91]。 然而,最近的研究表明,在BZY20 中使用燒結助劑,雖然可以提高BZY20 的燒結性,但是質子摻入能力和質子傳導能力顯著降低,同時空穴傳導增強,總體是不利于BZY20 的導電性能[92]。 摻雜通常涉及晶格缺陷,通過增加氧空位濃度來提高質子電導率,同時由于摻雜劑的影響,可以實現燒結性能的改善。 研究表明,使用In、Pr、Sn 與Y 對BaZrO3進行共摻雜能夠有效地提高BaZrO3的燒結性能同時不損害BaZrO3的化學穩定性[93-95]。 SUN 等人研究了Y 和In 共摻雜的BaZrO3,BaZr0.8Y0.2-xInxO3-δ。 隨著In 濃度的增加,BaZr0.8Y0.2-xInxO3-δ的燒結活性顯著提高,在濕H2中(3% H2O)600 ℃時BaZr0.8Y0.15In0.05O3-δ的總電導率最高可以達到1.6 mS/cm,對CO2、H2O 蒸氣和H2還原也表現出很高的化學穩定性[95]。 同時,SUN 等人以簡單的滴涂技術,于1 400 ℃空氣中反應5 h,在陽極襯底上成功地制備出了完全致密的BZYI5 電解質薄膜,BaZr0.8Y0.15In0.05O3-δ基單電池的功率密度在700 ℃時最高可達379 mW/cm2。 此外,一些新的制備工藝能夠有效地降低BaZrO3的燒結溫度,提高BaZrO3的性能。 KHANI 等人通過微乳液法以及丙烯酸酯水凝膠法制備了高度均勻的超細BaZr0.9Y0.1O2.95粉末,在1 500 ℃焙燒10 h 后致密度達到93%,600 ℃時的電導率為2 mS/cm[96]。 除了采用傳統的高溫燒結技術外,先進的薄膜制備技術對BaZrO3基電解質性能的提高起著重要作用。 PERGOLESI 等人采用PLD 在(100)取向的MgO 襯底上獲得了高度織構外延取向的BZY20 薄膜,由于沒有晶界的阻礙,BZY20 薄膜在500 ℃時質子電導率高達0.11 S/cm[97]。 2017 年,BAE 等人通過PLD 制備的BaZr0.85Y0.15O3-δ薄膜在組裝成單電池后,600 ℃時燃料電池的最大功率密度達到了740 mW/cm2[98]。 雖然通過PLD 能夠制備得到性能優異的BaZrO3基電解質,然而高昂的制造成本使其目前還無法大規模應用。
3.2.3 鋯酸鋇-鈰酸鋇基質子導體
BaCeO3具有優異的離子電導率以及良好的燒結性,BaZrO3具有良好的化學穩定性, 通過形成BaCexZr1-xO3固溶體能夠有效地整合二者的優點同時規避其缺點。 通過對BaCe0.9-xZrxY0.1O3-δ(BCZY)系統地研究發現,Zr 含量的增加可以提高BCZY 的化學穩定性,而Ce 含量的增加則提高了BCZY 的導電性以及可燒結性[99]。 為了改善BCZY 燒結性能,一般采用過渡金屬作為燒結助劑,NIKODEMSKI 等人研究了NiO、Fe2O3、WO3、PdO 等15 種金屬氧化物燒結助劑對BCZY 體系相形成和致密化的影響[100]。 研究結果表明,當添加的金屬離子半徑與Zr4+離子半徑相近而且具有穩定的+2 價時,能夠有效增加BCZY 的燒結性能,同時產生大量缺陷,對離子的傳導起著積極的促進作用;當金屬離子具有多種狀態,但是離子半徑與Zr4+離子半徑相近時,可以通過形成具有有限晶粒尺寸的機械穩定的高孔隙微結構來導致部分燒結;當金屬離子半徑遠離Zr4+離子半徑或者是穩定價態大于+2 價時,由于不能與BaZrO3形成固溶體,對燒結行為沒有改善作用。 因此常用的燒結助劑主要是NiO、ZnO、CuO 和CoO 四種。 NASANI 等人研究了NiO、ZnO 和CuO 作為燒結助劑對BCZY 性能的影響,相比于不添加燒結助劑,在1 400 ℃煅燒的BCZY的相對密度僅為86%,使用燒結助劑后相對密度提高到了95%以上,但是在所有情況下,添加燒結助劑均會顯著降低整體電導率,而晶界電導率相對不受影響[101]。
相比于使用燒結助劑會對BCZY 電解質的性能產生影響,一些特殊的燒結方式正在被積極研究。 比如WANG 等人通過微波燒結法制備得到晶粒尺寸僅為25 nm 的BCZY 粉末,在1 300 ℃燒結5 h 后相對密度達到了96%,而且以該電解質膜組裝的燃料電池在700 ℃功率密度達到了791 mW/cm2[102]。 此外,通過改進BCZY 粉體的制備方法也能有效提高電解質的性能。 FAN 等人以Tween-80 為表面活性劑,采用碳酸鹽共沉淀法制備了粒徑在150~300 nm 的BCZY 粉末,在1 400 ℃、無燒結助劑的情況下,BCZY粉末與陽極基板共燒結后,電解質高度致密化。 電解質厚度為20 μm、使用濕H2(3%H2O)作為燃料和環境空氣作為氧化劑的燃料電池700 ℃時的峰值功率密度高達1 050 mW/cm2[103]。
總體而言,BaCexZr1-xO3基質子導體表現出巨大的應用潛力,但是在電導率與穩定性之間的平衡仍然需要進一步研究。
3.3 鋯礦衍生的質子導體
由于獨立礦床較少,鋯礦是一種稀有金屬礦產資源。 目前鋯石、斜鋯石、異性石、鈉鋯石以及鋯鉭礦等是工業提取鋯的主要礦物。 由鋯礦加工而成的鋯產品應用廣泛,如碳酸鋯是紡織、造紙、涂料、化妝品行業的重要原料;氯氧化鋯可以用于紡織、皮革、橡膠添加劑、金屬表面處理劑、涂料干燥劑、耐火材料等;氧化鋯(ZrO2)適用于精密陶瓷、電子陶瓷、光學透鏡、玻璃添加劑、人造寶石、耐火材料、研磨拋光等行業;而金屬鋯更是重要的戰略金屬,主要用于核動力航空母艦、核潛艇和民用發電反應堆的結構材料、鈾燃料元件的包殼等。 然而我國作為全球最大的鋯資源消費國,2022 年鋯金屬儲量僅有7.16 萬t,需要大量依賴海外進口,因此鋯礦資源的高效利用是緩解鋯資源緊張的有效手段。 鋯基質子導體主要有3 類,分別是上文介紹過的螢石結構La2Zr2O7、鈣鈦礦結構的BaZrO3以及ZrO2,前2 種已經在上文進行了詳細介紹,這里就不在贅述。
ZrO2表現為螢石結構,Y 摻雜的ZrO2(YSZ)常作為SOFC 中的電解質,用以傳導O2-,隨后發現具有納米結構的YSZ 存在質子傳導而被廣泛研究。 在早期工作中,KIM 等人在潮濕環境下觀察到YSZ 納米晶中質子的傳導,且隨著YSZ 晶粒尺寸的減小質子電導率增加,室溫下,當晶粒尺寸從100 nm 減小到13 nm,質子電導率增加3 個數量級[104]。 他們認為晶界是質子傳輸路徑,并將導電性歸因于燒結時產生的高晶界密度。 2013 年,SCHERRER 等人研究了多孔YSZ 薄膜和致密YSZ 薄膜在低溫下的質子傳導[105]。 他們制備了具有不同微觀結構和晶粒尺寸的薄膜和塊狀樣品,以探索質子是否通過多孔表面或晶界傳輸。 在多孔膜中,室溫下發現了相當高的電導率,室溫至120 ℃,主要發生的是質子沿著YSZ 內表面物理吸附水進行傳導,由于其高度依賴于周圍大氣的濕度,因此隨著溫度的升高電導率降低。 在120~400 ℃的溫度范圍內,導電性被熱激活,活化能在0.4 eV 到1.1 eV 之間,導電是由O2-和H+所貢獻,H+傳導是由多孔膜內表面的羥基引起的。 大于400 ℃時,活性能為0.9 eV 至1.3 eV,O2-導電率占主導地位。在納米多孔體陶瓷YSZ 中也可以觀察到相同的行為,與納米多孔YSZ 相比,完全致密的納米晶體薄膜僅顯示出O2-導電性。 由于多孔膜和致密膜的主要區別在于孔隙率或者開孔的體積,而不是晶粒尺寸,因此可以得出結論,質子是沿著內部開孔的表面而不是晶界傳導的。 MIYOSHI 等人的研究也支持了這一結論,他們采用納米粉末合成與室溫超高壓壓實(4 GPa)相結合的新制備方法,制備了具有不同熱穩定性和遷移率的羥基和水分子分級結構的YSZ 致密納米晶顆粒[38]。 YSZ 樣品分別在中溫和低溫區域顯示出基于Grotthuss 機制和Vehicle 機制的高質子電導率。 表面終止羥基、氫鍵水分子和自由H2O 分子構成納米結構氧化物內的分級界面水合層,具有熱穩定性和同位素交換動力學的特征,這表明界面水合層在作為表面質子傳導途徑方面發揮著重要作用。 2017年,STUB 等人在較寬的相對濕度(RH)和溫度(25~400 ℃)范圍內研究了多孔YSZ 陶瓷樣品的電性能和載流子機制[39]。 多孔YSZ 在低于150 ℃時表現出純H+傳導,當相對濕度超過60%時,質子傳導機制從Grotthuss 機制向Vehicle 機制變化,與吸附水層從“類冰”結構向“類水”結構的變化同時發生。 同時吸附水層的厚度也會影響帶電物種的形成焓和遷移焓,當相對濕度從20%增加到84%時,質子傳輸的活化能從0.43 eV 降低到0.28 eV。 而SUN 等人的研究工作表明不同燒結溫度引起的結晶和優選晶面的差異可能對表面質子傳導產生重大影響[106]。 較低的燒結溫度使樣品形成相對圓形的非晶晶粒表面,從而有利于分子化學吸附,而高燒結溫度誘導形成優選的表面取向,導致強烈的解離化學吸附,從而導致高的活化能。
3.4 磷礦衍生的質子導體
磷礦石作為磷化工的重要原材料,其在工業上的應用已經超過100 a,我國磷礦石全球儲量第二,擁有36.90 億t 的儲量,其相關產品已經深入工業、農業、食品、醫藥、新能源等領域。 而通過磷化工加工得到的磷酸鹽是一類結構多樣的酸性固體,根據不同的金屬陽離子與磷酸根陰離子組合以及采用不同合成方法,制備得到的磷酸鹽結構可以從三維開放骨架到二維層狀網絡結構再到一維聚合物[107]。 磷酸鹽含有酸性磷酸基團(H2PO-4或),水分子可以在磷酸鹽骨架內形成氫鍵,因此磷酸鹽可作為質子導體。此外,磷酸鹽成本低廉,具有親水性、良好的熱穩定性、結構可設計性等優點。 磷酸二氫銫(CsH2PO4)、焦磷酸鹽、雜多酸、聚磷酸鹽等因具有優異的質子電導率而被廣泛研究。
3.4.1 磷酸二氫銫基質子導體
酸式鹽由堿金屬陽離子(Li+、K+、Rb+、Cs+)或者銨根離子與不完全電離的多元酸根陰離子組成,表現出介于正酸和鹽之間的性質。 酸式鹽在室溫下表現出較低的質子傳導性,但是當溫度升高到一定程度時,酸式鹽可以發生超質子相變,形成動態無序的氫鍵網絡,從而使得質子電導率提高4 ~ 5 個數量級[108]。 同時,由于酸式鹽本身可以形成氫鍵網絡,能夠在無水條件下傳導質子,而且具有良好的抗H2和O2滲透性能,可以提高PEMFC 的開路電壓,這些性質有助于PEMFC 在較高溫度下的運行[109]。 酸式鹽作為質子導體研究最多是硫酸氫銫(CsHSO4)、硒酸氫銫(CsHSeO4)以及CsH2PO4。
CsHSO4和CsHSeO4受熱會分解,被H2還原成H2S 或H2Se 氣體從而毒害催化劑,因此,CsH2PO4是研究最廣泛的一種酸式鹽。 CsH2PO4在230 ℃時發生超質子相變,相變后質子電導率從223 ℃時的8.5 μS/cm 提高到233 ℃時的18 mS/cm,同時在氧化/還原氣氛下表現出良好的穩定性,在Pt 催化作用下也沒有HxP 物種的生成[110]。 UDA 和HAILE 在不銹鋼氣體擴散電極上采用懸浮液沉積法制備了CsH2PO4厚度為25 μm 的燃料電池,開路電壓為0.98 V,240℃時功率密度達到了415 mW/cm2[111]。 然而CsH2PO4在250 ℃以上會脫水失重形成焦磷酸氫鹽和多磷酸鹽,直至完全分解成CsPO3[112]。 但是CsH2PO4的分解是可逆的,OTOMO 等人的研究表明,當環境中水分壓在0. 3 atm 及以上時能夠抑制CsH2PO4的分解并使CsH2PO4保持高的質子電導率[113]。
酸式鹽雖然發生超質子相變時具有高的質子電導率,但是酸式鹽的水溶性以及較差的機械性能使其在實際應用中仍然面臨困難。 通過在酸式鹽中加入SiO2、Al2O3、ZrO2等氧化物顆粒形成復合材料,酸式鹽與氧化物顆粒之間的相互作用可以產生非晶相,增加復合材料機械強度的同時在超質子相變溫度以下增強質子傳導[114-116]。 LEAL 等人通過機械研磨將CsH2PO4與SiO2進行復合,SiO2的加入提高了材料的穩定性,在270 ℃時添加SiO2的CsH2PO4比純CsH2PO4電導率高出一個數量級[116]。
3.4.2 焦磷酸鹽基質子導體
MP2O7(M=Sn、Ce、Ti、Zr)在100~300 ℃、低濕度或者干燥條件下具有優異的質子導電性(10-3~10-2S/cm),是最具潛力的中溫質子導體之一。 低價金屬離子(如In3+、Sc3+、Al3+、Mg2+)的摻入能夠有效地提高焦磷酸鹽的質子電導率,In0.1Sn0.9P2O7在250℃下的電導率高達0. 195S/cm,Mg2+的摻入將CeP2O7的電導率從 30 mS/cm 提高到 40 mS/cm[117-118]。 關于焦磷酸鹽高質子電導率的原因目前還存在爭議。 JIN 等人認為焦磷酸鹽中質子的摻入是H2O 與電子空穴作用形成,但是并未得到廣泛認同[119]。 此外,還有研究指出焦磷酸鹽高質子電導率可能與晶界或氣孔中次生的無定形相或吸水相有關,或者是合成過程使用過量的H3PO4前驅體和不充分的熱處理溫度,導致H3PO4在焦磷酸鹽表面殘留等[120-121]。
由于焦磷酸鹽難以形成致密的顆粒,因此以其組裝的燃料電池難以獲得較高的開路電壓,而且存在較大的氣體交叉[108]。 為了解決致密性問題,通常將焦磷酸鹽與聚合物進行復合,從而獲得較好的電池性能。 HEO 等人通過將90%的In0.1Sn0.9P2O7與有機磺酸進行混合制備出復合In0.1Sn0.9P2O7電解質材料,組裝成燃料電池后開路電壓為0.97 V,200 ℃下的功率密度可以達到187 mW/cm2[122]。 LEE 等人通過將SnP2O7與Nafion 混合制備出復合膜,通過在催化劑中加入季銨鹽系聚苯乙烯離聚體粘結劑,在240℃下獲得了870 mW/cm2的優異電池性能[123]。
3.4.3 磷鎢酸基質子導體
雜多酸也稱為多金屬氧酸,總共有6 種結構,其中Keggin 類型的雜多酸酸性更強,且結構穩定,化學式為HnXM12O40,X 為雜原子(P、Si),M 為多原子(W、Mo、V)。 其結構為中心的雜原子與氧形成四面體,多原子與氧形成八面體,12 個八面體圍繞在一個四面體周圍組成雜多酸陰離子[124]。 雜多酸分子通過形成H+、H3O+、H5O2+等離子來橋接水分子,這種水合的水分子結合松散,因此雜多酸表現出高質子傳導性[125]。 磷鎢酸、磷鉬酸在室溫下完全水合時,質子電導率可以達到0.18 S/cm。 然而雜多酸易溶于水,導致純雜多酸質子導體膜失效,可以通過浸漬法或是自組裝將雜多酸負載到介孔SiO2載體上[126-127]。與簡單的雜多酸與介孔SiO2共混相比,由于雜多酸陰離子錨定在介孔SiO2通道內,雜多酸的耐水性顯著提高,且隨著雜多酸含量的增加,雜多酸納米團簇之間的平均間距減小,降低了質子遷移的能壘,從而提高了質子的傳導性能。 通過將磷鎢酸真空浸漬到介孔SiO2上制備的復合電解質膜,25~90 ℃時質子電導率為0.07~0.11 S/cm,且降低了磷鎢酸對水的敏感性[128]。 而以磷鎢酸/介孔SiO2組裝的燃料電池在150 ℃下,沒有外部加濕的情況下功率密度達到372.1 mW/cm2,且電池運行8 d 輸出功率幾乎恒定[129]。
3.4.4 聚磷酸鹽基質子導體
聚磷酸銨(簡寫為NH4PO3)本身是不良質子導體,在干燥氛圍下250 ℃時,電導率僅為10-7S/cm,隨著溫度的升高,NH4PO3開始部分分解生成HPO3,電導率隨之急劇增加,300 ℃時質子電導率達到5 mS/cm[130]。 NH4PO3在濕潤氣氛下也表現出較高的質子電導率,在濕潤的空氣中,50~275 ℃溫度范圍內質子電導率可達0.012~2.61 mS/cm[131]。 為了提高NH4PO3的熱穩定性以及力學性能,通常將NH4PO3與(NH4)2MP4O13(M=Si、Ti、Sn)、焦磷酸鹽、氧化物(SiO2、TiO2、P2O5-SiO2)進行復合[132-135]。
4 結論與展望
礦物材料的開發利用為無機質子導體的發展提供了堅實的基礎。 同時無機質子導體的開發,拓寬了礦物材料的應用領域,提高了礦物材料的利用效率。在眾多類型的無機質子導體中,具有螢石結構、燒綠石結構以及鈣鈦礦結構的氧化物,擁有優異的質子傳導性能、良好的穩定性以及簡單的制備工藝,是目前無機質子導體的主要研究對象。 氧化物型質子導體主要面臨質子傳導性能和可燒結性的挑戰。 采用不同金屬離子對氧化物進行摻雜,通過增加氧空位濃度的方式來增加質子缺陷濃度,是提高氧化物質子傳導性能的關鍵。 摻雜離子的半徑和濃度影響著氧化物的整體性能,摻雜離子半徑過大或是過小均難以取代氧化物中活性位點上的離子產生氧空位。 摻雜濃度過低,氧空位產生較少,質子傳導性能提高不明顯;摻雜濃度過高,將會引起氧化物結構的不穩定。 因此,金屬離子摻雜存在最佳摻雜離子和摻雜濃度,從而導致離子摻雜對氧化物性能的提高存在上限。 為了提高氧化物的可燒結性,通常的策略就是添加燒結助劑,但是燒結助劑的加入會對氧化物的質子傳導性能產生不利的影響,如生成不導質子的雜相、導致摻雜離子的偏析等。 基于目前的研究現狀,在后續氧化物型質子導體的研究中可以從以下幾個方面考慮:
(1)氧化物界面工程的研究,界面作為質子在氧化物上傳導的重要路徑,通過對氧化物表面進行功能化改性是提高電解質性能的重要手段。
(2)復合型氧化物質子導體的研發,將氧化物與其他組分進行復合,通過調控不同組分之間的相互作用,從而獲得高性能質子導體。 如將氧化物與低溫熔鹽進行復合,利用熔鹽的高溫熔融的特性,在增強電解質致密性的同時構筑超質子傳導界面。
(3)新的低成本燒結技術的開發,放電等離子燒結、PLD、微波燒結等非常規燒結技術在提高燒結效率、降低能源消耗、改善材料性能等方面表現出巨大的潛力,人工智能的引入、新型燒結介質的開發以及先進的控制系統能夠有效降低這些燒結技術的成本。
