豬流行性腹瀉病毒YJH141分離株感染性克隆的構建及鑒定
2023-11-27 06:55:26鄭偉康張靜苗劉雅琦周瑩珊宋厚輝董婉玉王曉杜
中國獸醫(yī)學報 2023年9期
鄭偉康,張靜苗,劉雅琦,周瑩珊,杜 靜,宋厚輝,董婉玉,王曉杜
(浙江農林大學 動物科技學院/動物醫(yī)學院 浙江省畜禽綠色生態(tài)健康養(yǎng)殖應用技術研究重點實驗室/動物健康互聯(lián)網檢測技術浙江省工程研究中心/浙江省動物醫(yī)學與健康管理國際科技合作基地/中澳動物健康大數(shù)據(jù)分析聯(lián)合實驗室,浙江 杭州 311300)
豬流行性腹瀉病毒(porcine epidemic diarrhea virus,PEDV)是一種有囊膜單股正鏈RNA病毒,屬于套式病毒目(Nidovirales)、冠狀病毒科(Coronaviridae)、冠狀病毒屬(Coronavirus)的成員[1]。根據(jù)系統(tǒng)發(fā)育樹分析,冠狀病毒又可分為α、β、γ和δ 4個亞群,PEDV屬于α亞群,是典型的α冠狀病毒[2]。PEDV的基因組全長約為28 kb,5′端有帽子結構,3′端有poly(A)尾結構,病毒基因組全長包含7個開放閱讀框(open reading frame,ORF),分別編碼16個非結構蛋白(non-structure protein,Nsp),命名為Nsp1~16[3-4];4個結構蛋白,分別為刺突(spike,S)蛋白、囊膜(envelope,E)蛋白、膜(membrane,M)蛋白和核衣殼(nucleocapsid,N)蛋白;此外,還有一個由ORF3編碼的病毒復制過程中非必須的輔助因子[5]。
豬流行性腹瀉(porcine epidemic diarrhea,PED)在世界各地呈地方性流行趨勢,1971年首次暴發(fā)于英國,此后蔓延至歐洲多個國家;隨后,亞洲多個國家也相繼出現(xiàn)該病的流行,2007年泰國出現(xiàn)PED的大流行,越南則是在2009年暴發(fā)[6-8]。我國在1973年首次出現(xiàn)PED的流行,從20世紀80年代至今,PED在我國一直呈散發(fā)或地方性流行[9-10]。當前PEDV在我國豬病毒性腹瀉病中占主導地位,在我國豬群中PEDV的陽性率高達69.2%,其感染率明顯高于豬傳染性胃腸炎病毒(transmissible gastroenteritis virus,TGEV)和豬輪狀病毒(porcine rota virus,PoRV)[11-14]。同時,PEDV病毒還在不斷進化,有研究對2015—2018年分離的147份PEDV樣品與CV777毒株進行同源性比較后發(fā)現(xiàn),這些樣品與CV777毒株核苷酸同源性為90.7%~91.8%,氨基酸同源性僅為89.3%~90.7%,并且氨基酸突變概率在2015—2018年之間持續(xù)增加[15]。
近年變異毒株的接連出現(xiàn)使PED的流行呈現(xiàn)上升趨勢,并且發(fā)病情況也趨于復雜化,通常會出現(xiàn)不同毒株的交叉感染,這使得PEDV的防控未取得實質性的成效[16]。反向遺傳學操作技術可在基因水平對病毒進行改造,從而探究這些突變對病毒表型及性狀可能的影響,是研究病毒基因組結構和功能的重要工具。因此本研究利用反向遺傳學操作技術構建了流行毒株PEDV YJH141的全長感染性克隆,旨在為進一步深入研究該病毒基因組結構功能、復制及致病機理奠定基礎。
1 材料與方法
1.1 病毒與細胞系PEDV YJH毒株于2015年分離于浙江某豬場發(fā)病豬,體外連續(xù)傳代至141代,命名為YJH141(MT646162.1),保存于本實驗室。非洲綠猴腎細胞(Vero,C1008)及293T細胞購買于中國科學院上海細胞庫。
1.2 載體與菌株大腸桿菌E.coliDH10B、E.coliDH5α感受態(tài)細胞、pBAC載體質粒為本實驗室保存。
1.3 主要試劑及耗材反轉錄試劑盒購自寶日醫(yī)(TaKaRa)生物技術(北京)有限公司。PCR反應擴增酶KOD One PCR Master Mix-Blue、pMD18-T Vector試劑盒、DNA A-Tailing試劑盒購自東洋紡(ToYoBo)生物技術有限公司。限制性內切酶和T4DNA連接酶購自NEB生物技術(北京)有限公司。TRIZOL購自英濰捷基(上海)貿易有限公司。本研究中所用抗PEDV-N蛋白抗體 -為實驗室自制的鼠源抗體,熒光標記二抗為驢抗鼠,購自于英濰捷基(上海)貿易有限公司。所需的引物合成及測序服務均由杭州擎科生物科技有限公司提供。
1.4 YJH141全長cDNA克隆構建提取YJH141病毒液的RNA,反轉錄為cDNA,應用PCR將PEDV基因組分為8個片段進行分段擴增(圖1)。擴增片段分別克隆至T載體,然后將病毒不同片段克隆到pBAC載體,構建含有病毒完整基因組pBAC質粒。在引物設計過程中插入2個同義突變位點,使其在ORF1a末端構建1個SfiⅠ酶切位點,以此作為重組病毒的遺傳標記位點區(qū)別于親本病毒,引物信息見表1。

表1 全長感染性克隆構建引物信息
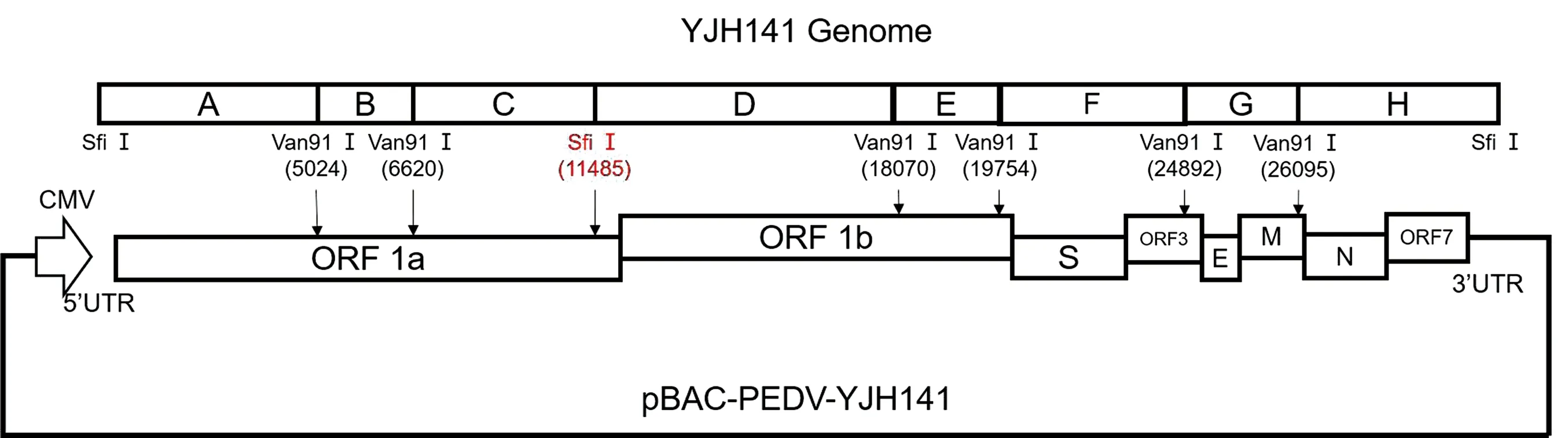
圖1 全長感染性克隆構建策略
1.5 YJH141全長cDNA克隆質粒的驗證將上述獲得的酶連產物轉化涂板后選取單克隆于LB培養(yǎng)基中進行培養(yǎng)。隨后設計針對片段連接處的引物對挑取的單克隆進行PCR擴增鑒定,引物信息見表2。將PCR鑒定陽性的克隆進行測序鑒定,正確的單克隆提取質粒命名為pBAC-PEDV-YJH141。
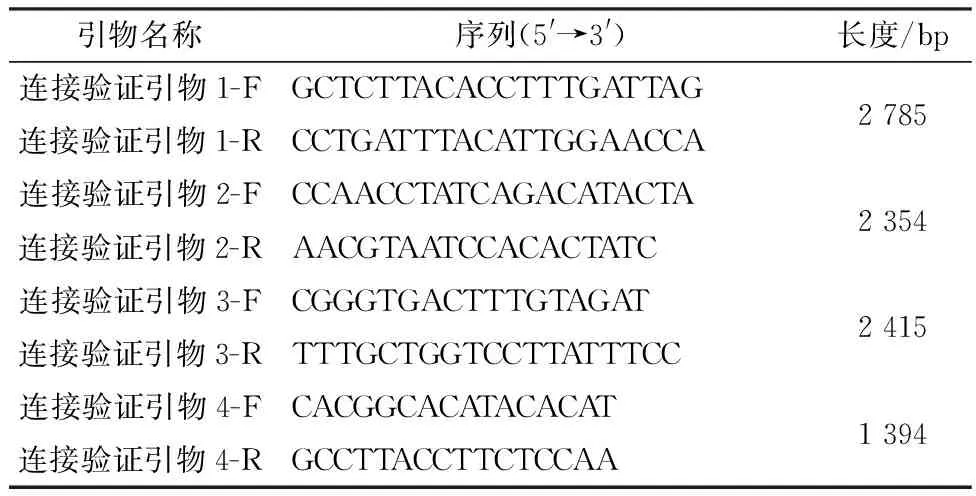
表2 全長感染性克隆驗證引物
1.6 病毒拯救及鑒定將質粒轉染至293T細胞(根據(jù)試劑盒說明操作)。轉染24~36 h后凍融1次收取上清接種于單層Vero細胞上。盲傳3代以確定病毒能夠穩(wěn)定傳代。通過觀察細胞病變(cytopathic effect,CPE)、RT-PCR驗證、遺傳標記位點確認及Western blot檢測等方法確認重組病毒拯救情況,并將拯救成功的病毒命名為rYJH141。
1.7 空斑試驗將Vero細胞鋪于6孔板,將待測病毒液用病毒維持液(胰酶終質量濃度為5 mg/L的DMEM培養(yǎng)基)10倍濃度稀釋后感染Vero細胞,每個稀釋度3個重復。置于37℃、5% CO2細胞培養(yǎng)箱孵育2 h,棄去培養(yǎng)上清,每孔加入2×DMEM(含10 mg/L胰酶)和2%低熔點瓊脂糖等體積混勻的培養(yǎng)基2 mL,室溫凝固后,置于37℃、5% CO2細胞培養(yǎng)箱中倒置培養(yǎng)48~72 h。隨后用4%多聚甲醛室溫固定2 h以上。棄去培養(yǎng)基,用結晶紫染液室溫染色30 min,清水輕柔洗去多余結晶紫染液,觀察空斑形成情況,分析其大小及形態(tài)差異。
1.8 間接免疫熒光檢測將待檢病毒以0.01 MOI感染Vero細胞,于18 h后棄去細胞培養(yǎng)液。加入4%多聚甲醛固定30 min。棄去多聚甲醛加入0.5% TritonX-100室溫透化細胞10 min。轉入含0.2%牛血清白蛋白(BSA)的PBS中37℃封閉1~1.5 h。以Mouse anti-PEDV-N(1∶800)為一抗,37℃孵育30 min,Donkey anti-mouse(1∶2 000)為二抗,37℃孵育45 min。最后利用DAPI復染劑(1∶1 000)室溫染核10 min。將細胞爬片取出滴加封片劑封片,利用激光共聚焦顯微鏡觀察。
1.9 病毒多步生長曲線測定Vero細胞鋪設于6孔板,置于培養(yǎng)箱中培養(yǎng)12~16 h,待細胞匯合度達到90%以上后棄去培養(yǎng)基,PBS潤洗2次。隨后以MOI為0.01待檢病毒感染6孔板中的Vero細胞,并分別于感染后6,12,18,24,36,48,60和72 h收取孔內細胞培養(yǎng)上清,每個時間點設置3個重復孔,按照Reed-Muench法計算病毒的TCID50,繪制病毒的生長曲線。
2 結果
2.1 pBAC-PEDV-YJH141重組質粒的構建及鑒定提取YJH141株的病毒RNA,通過反轉錄得到cDNA。按方法1.4操作方法,得到pBAC-PEDV-YJH141。通過PCR方法對全長質粒的各片段連接處進行鑒定,4對特異性引物均可擴增出預期大小的目的條帶,表明各片段按照預期成功連接(圖 2A~D)。選取2個驗證正確的單克隆進行質粒抽提并測序,結果表明正確構建了PEDV YJH141全長cDNA質粒,將其命名為pBAC-PEDV-YJH141(圖 2E)。
2.2 重組病毒rYJH141的拯救將pBAC-PEDV-YJH141轉染至293T細胞,收取293T培養(yǎng)上清接種于單層Vero細胞,同時以親本YJH141接毒組作為陽性對照,未接毒組作為陰性對照。在接種后12 h,2個接毒組鏡下觀察到明顯的CPE,18 h后兩者均產生大量合胞體,細胞出現(xiàn)崩解脫落;陰性對照細胞正常(圖3 A)。將病毒盲傳3次后,收取病毒培養(yǎng)上清提取重組病毒RNA,反轉錄為cDNA,利用特異性引物可擴增出N基因片段(圖3 B);同時收取細胞沉淀進行Western blot檢測,結果顯示重組病毒與親本YJH141均可檢測到N蛋白的表達(圖3 C)。通過PCR及測序驗證重組病毒的遺傳標記位點,結果顯示重組病毒帶有區(qū)別于親本病毒的同義突變位點(圖3 D)。上述結果均表明重組病毒拯救成功。
2.3 YJH141與rYJH141的空斑形成情況將重組病毒接種Vero細胞,以親本YJH141接毒組為陽性對照,未接毒組為陰性對照,于感染后48 h固定細胞并染色。結果顯示重組病毒與親本病毒形成的單個空斑面積相近,表明兩者在Vero細胞上產生細胞病變的能力接近(圖4)。
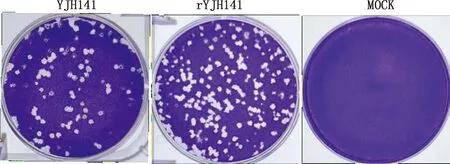
圖4 YJH141及rYJH141的空斑形成情況
2.4 YJH141與rYJH141的IFA檢測將重組病毒與親本YJH141病毒以0.01 MOI 分別感染Vero細胞,未接毒組為陰性對照,于18 h采用激光共聚焦試驗鑒定拯救的病毒。結果顯示YJH141與rYJH141組在感染后18 h時出現(xiàn)相似的熒光強度(圖5 C、F),而陰性對照組的細胞未出現(xiàn)紅色熒光(圖5 I),表明YJH141與rYJH141感染Vero細胞的能力接近。
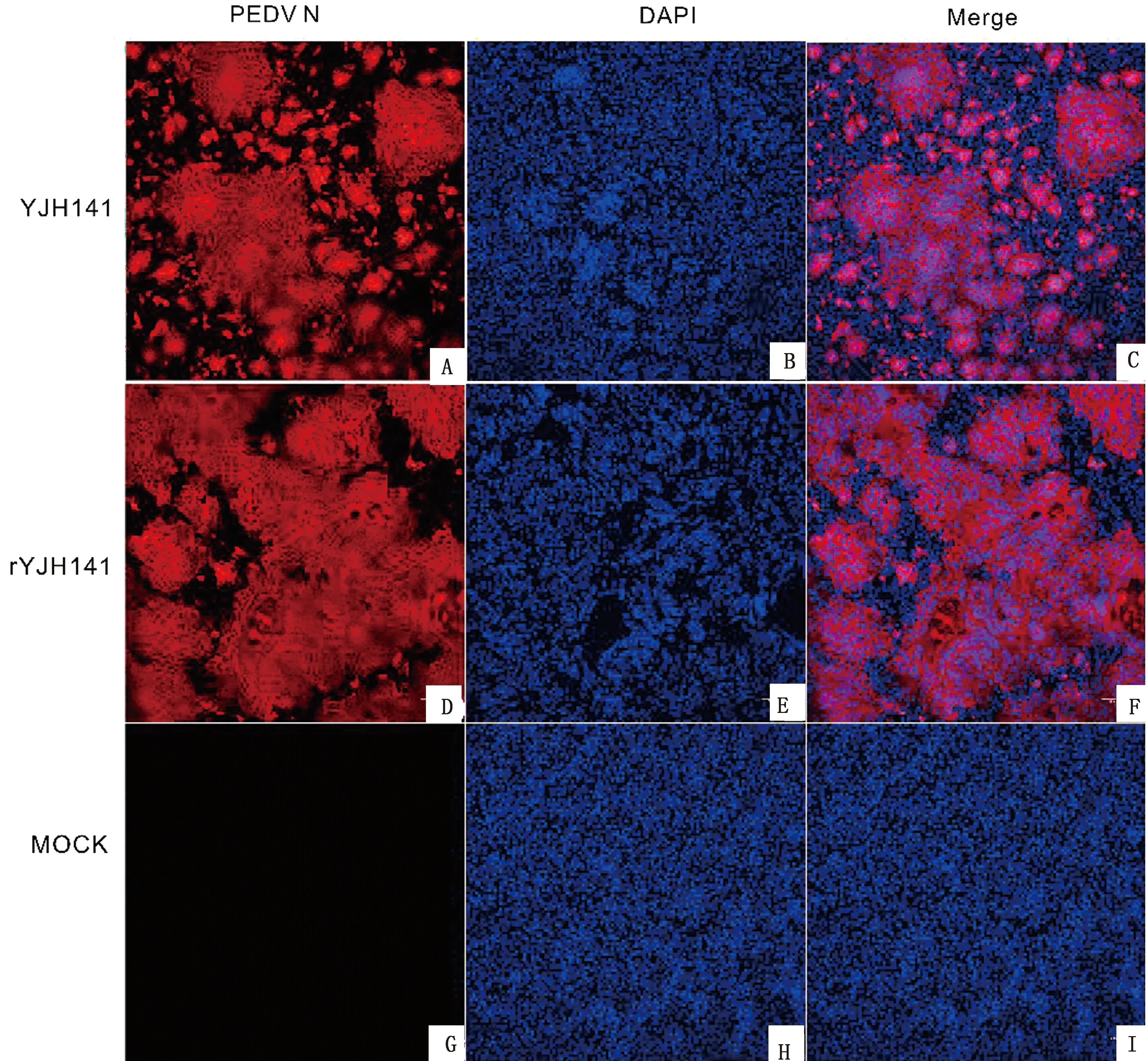
圖5 YJH141及rYJH141 N蛋白的IFA檢測
2.5 YJH141與rYJH141的生長曲線繪制將重組病毒與親本YJH141病毒以0.01 MOI接種于Vero細胞分別繪制病毒的多步生長曲線。結果顯示,rYJH141在Vero細胞上的多步生長曲線與親本YJH141相似,兩者無明顯差異(圖6)。表明YJH141與rYJH141呈現(xiàn)出相似的生長動力學特征。
3 討論
PEDV野毒株感染新生仔豬可引發(fā)極高的發(fā)病率和病死率,給養(yǎng)豬業(yè)造成了極大的危害[17]。PEDV屬于冠狀病毒α亞群,是典型的α冠狀病毒[2]。反向遺傳學操作系統(tǒng)是研究大部分RNA病毒特性的重要工具。冠狀病毒的反向遺傳學技術主要有靶向RNA重組技術、基于BAC載體的反向遺傳學操作技術以及基于體外連接的反向遺傳學操作技術等[18]。靶向RNA重組技術最早被應用于鼠肝炎病毒的拯救,該技術利用冠狀病毒在感染宿主細胞時RNA高效重組的特性,進而在病毒基因組中插入外源基因片段[19]。基于體外連接的反向遺傳學操作技術是通過獲得病毒全長cDNA,以全長cDNA為模板進行體外轉錄獲得用于翻譯病毒結構蛋白與非結構蛋白的RNA片段,將其電轉至細胞內翻譯包裝,從而獲得完整的病毒顆粒[18]。以上兩種方法都可以快速操作病毒基因組獲得重組病毒,但存在一定的局限性。靶向RNA重組技術只能對病毒基因組的一些特定片段進行編輯,并且重組病毒需要經過繁雜的純化過程,而基于體外連接的反向遺傳學操作技術需要進行體外轉錄,可能會由于轉錄過程中引入其他突變而導致拯救失敗。基于BAC載體的反向遺傳學操作系統(tǒng)可以有效彌補這些缺點。BAC載體的低拷貝以及可容納大片段插入的特性,使PEDV全長基因組可在BAC載體上穩(wěn)定保存,從而對病毒全長基因組的任意區(qū)段進行基因編輯,同時也避免了PEDV的復制酶基因片段不穩(wěn)定的問題[20]。
全長cDNA構建是構建病毒感染性克隆的關鍵,冠狀病毒是基因組最大的RNA病毒,長片段擴增會導致保真度降低,這給全長cDNA構建造成了較大的困難。因此,我們將病毒基因組分成8個節(jié)段進行擴增,同時使用PfuⅡ高保真的DNA聚合酶,這樣可高效擴增PEDV YJH片段且能夠有效避免突變的發(fā)生。此外,由于293T細胞擁有高效的轉染效率,我們采用先轉染293T細胞,收取細胞培養(yǎng)上清再感染PEDV易感細胞Vero的策略拯救PEDV流行毒株YJH141,成功建立了流行毒株PEDV YJH的反向遺傳操作平臺,獲得了感染性拯救病毒YJH141。通過IFA、空斑和生長曲線測定驗證了該拯救病毒與親本病毒在生物學特性方面保持一致。但由于全長感染性克隆質粒較大,在全長基因組上進行點突變或結構區(qū)域的突變需要先在小片段上進行編輯,然后所有片段進行酶切和連接,操作較為繁瑣,因此我們可以通過向該體系內引入CRISPR-Cas9系統(tǒng)高效編輯PEDV全長基因組。通過向導RNA的設計可以使Cas9成為一種可編程的核酸內切酶,CRISPR-Cas9系統(tǒng)可用于精準的大片段基因敲除和插入,可于體外高效編輯全長感染性克隆質粒,有助于提高克隆效率[21-22]。
反向遺傳學技術是研究病毒分子遺傳學十分重要的技術平臺。通過反向遺傳學探究PEDV的生物學特性也是近年來的研究熱點。有研究將PEDV毒株FJzz1在Vero細胞中連續(xù)傳代200次后發(fā)現(xiàn),在其S1蛋白的N端發(fā)生了連續(xù)的氨基酸缺失,并且該毒株的毒力有明顯下降[23]。另外,SATO等[24]將PEDV毒株83P-5在Vero細胞傳代至100代時S蛋白發(fā)生的13個氨基酸突變與DR13細胞適應株S蛋白的氨基酸突變基本相同。以上研究提示PEDV S蛋白中氨基酸的突變可能是導致PEDV適應細胞及毒力變化的關鍵分子基礎。這些毒株在傳代過程中出現(xiàn)的相關位點突變是否直接決定了其毒力及細胞適應性的變化需要通過反向遺傳學技術進一步驗證。綜上所述,本研究以BAC系統(tǒng)為基礎,成功構建了PEDV流行毒株YJH141的反向遺傳學操作系統(tǒng),為后續(xù)探究該病毒復制及致病機制的相關研究奠定了堅實基礎,也將為弱毒疫苗的研制提供平臺。
