芝麻酚通過AMPK信號通路調控巨噬細胞極化減輕脂肪組織炎癥
2023-11-10 13:31:58程明慧張勸勸
中國藥理學通報 2023年11期
關鍵詞:小鼠
劉 矚,程明慧,張勸勸,秦 虹
(中南大學湘雅公共衛生學院營養與食品衛生學教研室,湖南 長沙 410078)
隨著生活水平的提高,肥胖患者數量日益增加。研究表明,慢性炎癥是Ⅱ型糖尿病、高血脂及心血管疾病等多種肥胖相關代謝性疾病發病的關鍵因素[1]。脂肪組織作為與肥胖最密切相關的器官,是慢性炎癥的主要來源。因此,開發減輕脂肪組織炎癥的藥物對防治肥胖及其相關代謝性疾病具有重要意義。
巨噬細胞是慢性炎癥中浸潤脂肪組織的主要免疫細胞,在脂肪組織炎癥的發生發展中扮演核心角色[2]。巨噬細胞根據微環境不同可極化為促炎M1型和抗炎M2型。健康小鼠的脂肪組織巨噬細胞主要表現為M2型,而在高脂飲食誘導的肥胖小鼠脂肪組織中,巨噬細胞發生M1型向M2型的轉化,M1型巨噬細胞基因大量表達[3]。M1型巨噬細胞是炎癥因子的主要來源,促進炎癥的發生發展;M2型巨噬細胞分泌抗炎因子,拮抗炎癥反應[4]。調控巨噬細胞M1/M2型極化正成為治療多種炎癥相關代謝性疾病的新型策略[5]。腺苷酸激活蛋白激酶(AMP-activated protein kinase,AMPK)是一種酶復合體,在巨噬細胞極化和炎癥發生發展中都發揮重要作用。有研究提示,激活AMPK信號通路不僅可抑制肥胖小鼠脂肪組織中巨噬細胞的浸潤、減輕炎癥反應,還可抑制巨噬細胞M1型極化[6-7]。因此,尋找可激活巨噬細胞AMPK的藥物,將為防治炎癥相關代謝性疾病提供新的策略。
芝麻酚(sesamol,SEM)是一種從芝麻中提取的具有一個酚環結構的小分子天然木脂素,具有廣泛的藥理作用,如可通過抑制小膠質細胞中離子鈣結合銜接分子1的激活緩解小鼠神經炎癥[8]、可通過抑制IKKα/NF-κB信號通路減輕腎臟炎癥并阻止巨噬細胞浸潤[9]等。因此,SEM在減輕脂肪組織關炎癥方面可能也具有較高的藥用價值,值得深入探索。本課題組前期研究證實,SEM可激活肝臟和骨骼肌中的AMPK蛋白[10-11],顯著減輕肥胖相關病理反應。目前,SEM對肥胖相關脂肪組織炎癥的影響及作用機制尚不清楚,我們推測SEM可能通過激活AMPK通路調控巨噬細胞極化減輕脂肪組織炎癥。本研究首先利用高脂飲食(high fat diet,HFD)誘導肥胖小鼠模型,觀察到SEM可減輕脂肪組織炎癥反應,再體外采用脂多糖(lipopolysaccharide,LPS)建立炎癥細胞模型,探討SEM對巨噬細胞極化的具體調控機制,為SEM的抗炎作用提供理論依據和實驗基礎。

Fig 1 The chemical structure of sesamol
1 材料與方法
1.1 實驗動物及細胞8周SPF級雄性C57BL/6J小鼠購于湖南斯萊克景達實驗動物有限公司;RAW264.7細胞系購于中國科學院細胞庫(TCM13)。
1.2 實驗材料SEM(S3003,美國Sigma公司);普通小鼠飼料(D12450J,北京科澳協力飼料有限公司,含10%脂肪),高脂小鼠飼料(D12492J,北京科澳協力飼料有限公司,含60%脂肪);LPS(L2880,美國Sigma公司),compound C(HY-13418A,美國MCE公司),BCA試劑盒(南京建成公司),白細胞介素-6(interleukin-6,IL-6)ELISA試劑盒(ml00285,上海酶聯生物科技有限公司),白細胞介素-10(interleukin-10,IL-10)ELISA試劑盒(ml063159,上海酶聯生物科技有限公司)、單核細胞趨化因子-1(monocyte chemotactic protein,MCP-1)ELISA試劑盒(ml037840,上海酶聯生物科技有限公司)、腫瘤壞死因子-α(tumor necrosis factor alpha,TNF-α)ELISA試劑盒(ml002095,上海酶聯生物科技有限公司),兔抗p-AMPK(AF3423,Affinity)和AMPK(ER62660,杭州華安生物技術有限公司),核因子κB(nuclear factor kappa-B,NF-κB)(et1603-12,杭州華安生物技術有限公司),過氧化物酶體增殖物激活受體γ共激活劑-1α(peroxisome proliferator-activated receptor gamma coactivator -1alpha,PGC-1α)(A19674 ,ABclonal),CD11c(cluster of differentiation 11c,CD11c)(DF7583,Affinity)、CD206(cluster of differentiation 206,CD206)(DF4149,Affinity),GAPDH(glyceraldehyde-3-phosphate dehydrogenase,GAPDH)(ET1601-4,杭州華安生物技術有限公司)。
1.3 主要儀器Azure 600多功能熒光成像系統(美國Azure Biosystems),全波長酶標儀(美國 Thermo Fisher Scientific);超高速冷凍離心機(美國 Sci Logex)。
1.4 動物分組及干預將42只8周齡雄性C57BL/6J小鼠置于20~24 ℃,40%~60% 濕度,12 h光/暗交替循環的環境條件下喂養,自由進食和飲水。適應性喂養1周后隨機將其中12只小鼠作為對照組(CON組),喂養普通小鼠飼料,其余小鼠喂養高脂小鼠飼料。13周后肥胖模型造模成功(以超過對照組小鼠平均體質量的20%作為成模標準),將小鼠隨機分為高脂組(HFD組)和SEM干預組(SEM組),每組12只。CON組和HFD組每天給予 0.5%羧甲基纖維素鈉灌胃,SEM組每天給予SEM(0.5%羧甲基纖維素鈉混懸配制)灌胃,劑量為100 mg·kg-1,8周后,摘眼球取血,頸椎脫臼處死小鼠,剖取附睪脂肪組織樣本于-80 ℃冰箱分裝保存。實驗方案通過中南大學湘雅公共衛生學院倫理委員會審批(批準文號:XYGW-2021-75)。
1.5 細胞培養將RAW264.7巨噬細胞接種于RPMI 1640培養基(含10%胎牛血清),置于37 ℃、5%CO2條件下培養。待細胞融合度近70%時采取如下處理:空白對照組(CON組,RPMI 1640培養基培養)、模型對照組(LPS組,100 μg·L-1LPS處理24 h)、SEM低劑量(LPS+6.25 μmol·L-1SEM組,100 μg·L-1LPS處理24 h,并用 6.25 μmol·L-1SEM干預)、SEM高劑量干預組(LPS+25 μmol·L-1SEM組,100 μg·L-1LPS處理24 h,并用25 μmol·L-1SEM干預)、AMPK抑制劑組(LPS+25 μmol·L-1SEM+CC組,預先用AMPK抑制劑compound C 5 μmol·L-1處理2 h,余同LPS+25 μmol·L-1SEM組)。
1.6 MTT法測定細胞毒性作用取對數生長期RAW264.7巨噬細胞,以每孔2×104個的密度接種于96孔板,培養24 h后棄上清,并分別設置空白調零組、正常對照組和給藥組。給藥組包括3組藥物,第1組加入SEM(3.125~100 μmol·L-1),第2組加入LPS(12.5~400 μg·L-1),第3組加入適宜濃度LPS及SEM(3.125~100 μmol·L-1),每個濃度設置5個復孔,培養結束后每孔加入10 μL MTT溶液(5 g·L-1),于培養箱繼續孵育4 h后,輕輕棄去上清,每孔加入150 μL二甲基亞砜,置于搖床上振蕩使甲瓚結晶充分溶解,采用酶標儀于 490 nm波長處檢測各孔吸光度值,并計算細胞存活率。

1.7 ELISA法檢測趨化因子、炎癥因子和抗炎因子表達將血液于室溫凝固20 min,4 ℃ 3 000 r·min-1離心20 min,收集血清;將附睪脂肪組織(epididymal adipose tissue,eWAT)和 PBS以1 mg ∶9 μL(m組織∶VPBS)的比例制成脂肪組織勻漿,4 ℃ 3 000 r·min-1離心20 min,收集上清;收集RAW264.7巨噬細胞培養液,4 ℃ 3 000 r·min-1離心20 min,收集細胞上清。將上述樣品按照MCP-1、IL-6、TNF-α、IL-10試劑盒說明書進行濃度測定。
1.8 Western blot檢測相關蛋白表達以磷酸化蛋白酶抑制劑PMSF ∶RIPA裂解液=2 ∶1 ∶97的比例配置裂解液,裂解附睪脂肪組織和干預結束后的 RAW264.7巨噬細胞。采用BCA法檢測蛋白濃度后進行蛋白變性。上樣后電泳完成蛋白分離,將蛋白濕轉于NC膜上,并用5%脫脂牛奶于室溫下封閉1 h,敷一抗后于4 ℃ 冰箱過夜,洗膜后敷二抗于37 ℃ 孵育1 h,經ECL化學發光并用顯影儀進行顯影,應用ImageJ軟件分析附睪脂肪組織中CD11c及RAW264.7巨噬細胞中的AMPK、p-AMPK、PGC-1α、NF-κB、CD11c、CD206蛋白的相對表達量,用GraphPad Prism 8.0作圖。
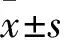
2 結果
2.1 SEM減輕肥胖小鼠炎癥反應炎癥反應中,常伴隨大量炎癥因子的產生。因此,我們應用ELISA法檢測各組小鼠血清及附睪脂肪組織中的炎癥因子IL-6、TNF-α和抗炎因子IL-10的分泌水平。如Fig 2所示,與 CON組相比,HFD組血清和附睪脂肪組織中的炎癥因子IL-6和TNF-α分泌均明顯上升,抗炎因子IL-10明顯下降;而與HFD組相比,SEM組中的IL-6、TNF-α分泌水平明顯降低,IL-10分泌卻有明顯增多。這表明,SEM能夠減輕肥胖小鼠炎癥反應。

Fig 2 Inflammation of obese mice alleviated
2.2 SEM抑制肥胖小鼠脂肪組織巨噬細胞M1型極化巨噬細胞作為脂肪組織中占比最多的免疫細胞,與脂肪組織炎癥的發生發展有著密切關系。為了明確SEM的抗炎機制,我們檢測了其對趨化因子MCP-1分泌水平、脂肪組織巨噬細胞中M1型極化標志物CD11c蛋白表達的影響。如Fig 3A所示,與CON組相比,HFD組中的小鼠血清和附睪脂肪組織中巨噬細胞趨化因子 MCP-1升高,而SEM干預后,MCP-1明顯下降。MCP-1的分泌與M1型巨噬細胞的激活密切相關,因此,我們檢測了脂肪組織中M1型巨噬細胞標志物CD11c的含量。如Fig 3B所示,與CON組相比,HFD組中的CD11c蛋白含量明顯增加;與HFD組相比,SEM組的CD11c蛋白明顯降低。這提示,SEM通過抑制脂肪組織中巨噬細胞M1型極化發揮抗炎作用。

Fig 3 M1 polarization of adipose tissue macrophages
2.3 SEM及LPS對RAW264.7巨噬細胞的毒性作用為了驗證推測,我們利用RAW264.7巨噬細胞進一步探索SEM抗炎作用的機制。首先,我們檢測了SEM、LPS對RAW264.7巨噬細胞的細胞毒性作用。MTT結果如Fig 4A所示,SEM以不同濃度作用RAW264.7巨噬細胞24 h后,在3.125~50 μmol·L-1時不影響RAW264.7巨噬細胞活性。如Fig 4B所示,用不同濃度LPS作用于RAW264.7巨噬細胞24 h后,細胞活力與對照組相比均無顯著性差異,我們選擇100 μg·L-1LPS 進行后續實驗。接下來,我們檢測SEM與100 μg·L-1LPS同時孵育時對RAW264.7巨噬細胞的細胞毒性作用,如Fig 4C所示,SEM濃度小于50 μmol·L-1時,RAW264.7巨噬細胞活性與對照組相比差異無統計學意義。因此,后續實驗采用100 μg·L-1LPS及6.25 μmol·L-1、25 μmol·L-1SEM進行。
2.4 SEM可激活巨噬細胞AMPK信號通路AMPK參與調控巨噬細胞炎癥反應及極化,我們之前的研究證明,SEM可調節肝臟和骨骼肌中的AMPK,因此推測SEM可能通過激活巨噬細胞AMPK調控巨噬細胞極化從而發揮抗炎作用。我們利用LPS刺激的RAW264.7巨噬細胞誘導出炎癥細胞模型,檢測細胞中AMPK、p-AMPK的蛋白含量。如Fig 5A所示 ,LPS抑制了RAW264.7細胞中p-AMPK的表達,而SEM可明顯減輕這種抑制,說明SEM可能對巨噬細胞AMPK信號通路有激活作用。接下來,我們檢測了 AMPK下游通路蛋白NF-κB和PGC-1α的蛋白含量,并利用AMPK抑制劑 compound C進行驗證。結果如Fig 5B所示,與空白對照組(CON組)相比,炎癥模型組(LPS組)細胞中AMPK下游蛋白PGC-1α蛋白量下調,NF-κB明顯上調;而較LPS組而言,6.25 μmol·L-1和 25 μmol·L-1的SEM干預均能使 NF-κB 蛋白量降低,并且 25 μmol·L-1SEM還能促進 PGC-1α 明顯升高;在給予 AMPK 抑制劑 compound C后,SEM對巨噬細胞 AMPK信號通路相關蛋白的調控作用明顯減弱。結果表明,SEM對巨噬細胞 AMPK信號通路具有激活作用。
2.5 SEM調控巨噬細胞極化與AMPK信號通路相關為了探究SEM調控巨噬細胞極化的效果和機制,我們檢測了M1型巨噬細胞極化標志物CD11c和M2型巨噬細胞極化標志物CD206蛋白含量。如Fig 6A所示,與Control組相比,LPS組CD11c有明顯升高、CD206明顯下降;與LPS組相比,SEM劑量依賴性降低巨噬細胞中的CD11c,提高 CD206蛋白量;而給予AMPK抑制劑后,SEM的上述作用被逆轉。這說明SEM能夠抑制巨噬細胞的M1型極化、促進M2型極化,并且AMPK信號通路在這個過程中發揮了重要作用。接下來,我們采用ELISA法檢測M1型巨噬細胞分泌的促炎因子IL-6、TNF-α和M2型巨噬細胞分泌的抗炎因子IL-10的水平。結果如Fig 6B所示,LPS作用巨噬細胞后,IL-6和TNF-α水平上升、IL-10水平下降;在給予不同劑量的SEM后,IL-6和TNF-α均下降,其中 25 μmol·L-1SEM的作用效果更好,并且能使IL-10有顯著上升;而AMPK抑制劑逆轉了SEM的作用效果。這進一步說明SEM可通過激活AMPK信號通路,調控巨噬細胞M1/M2型極化,減輕炎癥反應。
3 討論
肥胖所伴隨的慢性炎癥是Ⅱ型糖尿病、高血壓、高血脂等代謝性疾病發病的關鍵因素。脂肪組織是與肥胖癥密切相關的器官,脂肪組織炎癥在肥胖相關代謝性疾病的發生發展中起著十分重要的作用。

Fig 4 Effects of sesamol and LPS on cell viability of RAW264.7

Fig 6 Macrophage polarization regulated by sesamol through AMPK signaling
本研究發現,SEM可通過調節巨噬細胞極化降低肥胖小鼠脂肪組織炎癥水平,其分子機制可能是AMPK信號通路的激活。
巨噬細胞作為脂肪組織中占比最大的免疫細胞,在脂肪組織炎癥反應中扮演關鍵角色。巨噬細胞可因微環境改變而轉換為M1和M2兩種亞型,M1型巨噬細胞高表達CD11c,釋放大量炎癥因子加重炎癥反應,如IL-6、TNF-α;M2型巨噬細胞高表達CD206,可分泌抗炎因子抑制炎癥發展,如IL-10[4]。檢測這些特異性標志物是鑒別M1和M2巨噬細胞的常用方法。我們的研究結果發現,SEM減輕了HFD小鼠的炎癥狀態,主要反映在SEM干預后,小鼠血清和附睪脂肪組織中炎癥因子IL-6、TNF-α的降低以及抗炎因子IL-10的增加,這可能與SEM對巨噬細胞M1/M2型的調控相關。健康狀態下,脂肪組織中巨噬細胞主要表現為M2型,而肥胖患者和肥胖小鼠脂肪組織中的巨噬細胞主要呈M1型[12],活化的 M1巨噬細胞可分泌促炎因子激活炎癥級聯反應,形成正反饋通路,增強炎癥反應。我們經過對脂肪組織中M1型巨噬細胞標志蛋白CD11c的測定,證實SEM抑制了脂肪組織巨噬細胞的M1型極化。此外,活化的 M1型巨噬細胞還可分泌趨化因子(如MCP-1)募集更多血液中的單核細胞浸潤脂肪組織炎癥部位[2],進入正反饋通路,放大炎癥反應,加重組織損傷。因此,巨噬細胞浸潤的減少,也是M1型巨噬細胞極化減少的標志之一。本研究中,我們發現,SEM明顯降低血清與附睪脂肪組織中的MCP-1表達,進一步說明SEM抑制了肥胖小鼠脂肪組織中巨噬細胞的浸潤、減少了M1型極化,并減輕了炎癥級聯反應。這與Hsu等[13]的研究結果相似,他們通過大鼠應激性潰瘍模型證明了SEM減輕胃潰瘍的機制之一是抑制巨噬細胞浸潤胃黏膜組織。因此,我們通過建立高脂誘導的肥胖小鼠模型,觀察SEM對小鼠血清、附睪脂肪組織中炎癥相關因子、趨化因子及脂肪組織中M1型巨噬細胞標志物的影響,認為SEM可能調控巨噬細胞極化,減輕脂肪組織炎癥。
AMPK是一種能量感應絲氨酸/蘇氨酸蛋白激酶,近期有研究表明,AMPK信號通路的激活是促進巨噬細胞M1型向M2型極化和緩解炎癥的關鍵因素[6,14]。我們先前的研究證明,SEM可激活肝臟和骨骼肌中的AMPK[10-11],因此,推測SEM可能通過激活AMPK信號通路來調控巨噬細胞極化從而發揮抗炎效應。本實驗觀察到,SEM增強了炎癥狀態下巨噬細胞p-AMPK的表達,激活了AMPK蛋白。有研究提示,AMPK發揮調控巨噬細胞極化作用的機制可能是通過上調PGC-1α的表達、降低NF-κB的表達[15-16]。PGC-1α是一種轉錄共激活因子,可降低炎癥因子的產生,同時它也是線粒體功能的主要調節因子,可通過激活線粒體功能增強巨噬細胞氧化磷酸化,促進巨噬細胞向M2型極化[17]。NF-κB也被證實是一種十分重要的炎癥反應通路蛋白,是M1型巨噬細胞激活過程的關鍵轉錄因子之一,降低NF-κB可抑制M1 型巨噬細胞極化[18]。通過本研究我們發現,SEM可以增加巨噬細胞中PGC-1α蛋白、減少NF-κB蛋白表達,而AMPK的特異性抑制劑能夠逆轉SEM的這些作用。這進一步驗證了SEM對巨噬細胞AMPK 信號通路的激活作用。并且,炎癥狀態下的巨噬細胞被施與AMPK抑制劑后,SEM降低CD11c、 IL-6、TNF-α,增加CD206、IL-10水平等作用均被逆轉。因此,我們的實驗結果證明了SEM可以通過激活AMPK信號通路,調節巨噬細胞M1/M2極化以發揮抗炎作用。
綜上所述,本研究利用肥胖小鼠模型和炎癥細胞模型,發現SEM減輕脂肪組織炎癥反應與其對巨噬細胞極化的調控密切相關,其分子機制可能是激活了AMPK信號通路。本研究可為SEM在減輕肥胖相關炎癥的臨床應用上提供理論基礎,為SEM作為抗炎藥物的研發提供幫助。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
東方少年·布老虎畫刊(2023年8期)2023-08-01 15:45:12
科學大眾(2021年6期)2021-07-20 07:42:44
科學(2020年3期)2020-11-26 08:18:30
學苑創造·A版(2020年9期)2020-10-13 09:41:02
娃娃樂園·綜合智能(2019年3期)2019-04-03 09:17:36
中成藥(2018年2期)2018-05-09 07:19:34
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
湖北師范大學學報(自然科學版)(2015年2期)2016-01-10 08:41:55
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
