利用改進的Pictet-Spengler反應合成多取代β-咔啉
2023-10-31 07:39:00張京玉楊懷霞
合成化學 2023年10期
關鍵詞:催化劑
鐘 錚, 張京玉, 密 霞, 楊懷霞
(河南中醫藥大學 藥學院,河南 鄭州 450046)
β-咔啉是咔啉類化合物重要的代表性結構,廣泛存在于天然產物與生物活性小分子中,具有抗腫瘤、抗病毒以及中樞神經調節等重要生理活性[1-5]。常見的多取代β-咔啉類化合物的合成路線有2條:(1)首先合成取代的4-(2-硝基苯基)-吡啶前體,再以亞磷酸、三苯基膦等為還原劑,采用Codogan反應合成,或利用烏爾曼反應、氮烯插入反應合成[6-11]; (2)以多取代色氨為原料,經Pictet-Spengler反應得到四氫咔啉后,再經氧化脫氫得到產物[12-14]。第1條路線的優點是能夠更高效地在苯環上選擇性引入取代基,之后再與吡啶環通過Suzuki反應偶聯。但Suzuki反應中需要使用0價鈀催化劑,因而會使反應操作復雜,成本較高,不利于大規模制備。與第1條路線相比,第2條路線更為簡潔,不需要使用0價鈀催化劑,因而大多數文獻選擇采用第2條路線合成β-咔啉類衍生物。
經典的Pictet-Spengler反應通常在強質子酸如濃硫酸、對甲苯磺酸催化下完成,當反應底物中存在對質子酸敏感的基團時就無法采用該方法,因此尋找合適的非質子性Lewis酸催化劑成為該反應的主要改進方向之一。不過常見的Lewis酸并不適用于Pictet-Spengler反應,原因可能是反應產物抑制了Lewis催化劑活性。目前此類報道不多,偶有硫脲或磷酰胺類小分子催化劑用于該反應[15]。 MATTHEW等[16]報道了在制備四氫異喹啉類化合物時,以六氟異丙醇鈣為催化劑代替強酸,利用Pictet-Spengler反應合成苯并哌啶環結構類似物并取得55%~86%收率的結果。
為探索能夠在非酸性條件下利用Pictet-Spengler反應合成β-咔啉類化合物的新方法,本文以色氨類化合物與醛類為原料,六氟異丙醇鈣為催化劑,二氯甲烷(DCM)為溶劑,通過改進的Pictet-Spengler反應合成4個四氫-β-咔啉類化合物(圖1,1a~1d),再以2,3-二氯-5,6-二氰對苯醌(DDQ)為氧化劑脫氫芳構化得到多取代β-咔啉衍生物(2a~2d),其結構經1H NMR和ESI-MS確證。在關鍵步驟Pictet-Spengler反應中,為了確定反應的最優條件,考察了底物物料比、催化劑用量、溶劑種類和反應時間對收率的影響。
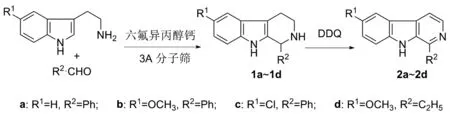
圖1 多取代β-咔啉的合成路線
1 實驗部分
1.1 儀器與試劑
TEMP-MELT型熔點儀;Bruker AMX-400型核磁共振儀(DMSO-d6溶劑,TMS內標);HP5989A型質譜儀。
六氟異丙醇鈣參考文獻自制[17],其余所用試劑均為市售分析純或化學純。
1.2 合成
(1)1a~1d的合成(以1a為例)
氮氣保護下,在反應瓶中依次加入六氟異丙醇鈣151 mg(0.4 mmol),色胺320 mg(2.0 mmol), 3A分子篩1 g(4.9 mmol),苯甲醛0.3 mL(3.0 mmol)和DCM 20.0 mL,攪拌使其溶解,室溫下反應24 h。加入0.5 mL飽和NaHCO3水溶液攪拌10 min后加入無水硫酸鈉干燥。過濾后將濾液減壓濃縮,殘余物經硅膠柱層析(洗脫劑:V二氯甲烷∶V甲醇∶V氨水=20.0 ∶1.0 ∶0.2)純化得灰白色粉末1a411 mg,產率83%。以5-甲氧基色胺、5-氯色胺代替色胺,丙醛代替苯甲醛,用類似方法合成灰白色粉末1b~1d。
1a: m.p.168~170 ℃(m.p.168~169 ℃[18]);1H NMRδ: 11.71(s, 1H), 7.87~7.84(dd,J=6.6 Hz, 1.8 Hz, 1H), 7.58~7.55(dd,J=7.2 Hz, 1.8 Hz, 1H), 7.38~7.22(m, 5H), 6.97~6.86(m, 2H), 5.58(s, 1H), 3.32~3.23(m, 2H), 2.71~2.67(m, 2H), 1.96(s, 1H); MS(ESI)m/z: 249.1{[M+H]+}。
1b: m.p.181~183 ℃;1H NMRδ:11.69(s,1H), 7.37~7.22(m, 6H), 7.12(s, 1H), 6.92~6.89(dd,J=7.3 Hz, 1.9 Hz, 1H), 5.57(s, 1H), 3.85(s,3H), 3.34~3.26(m, 2H), 2.71~2.67(m, 2H), 1.94(s, 1H); MS(ESI)m/z: 279.1{[M+H]+}。
1c: m.p.186~188 ℃;1H NMRδ:11.73(s,1H), 7.95~7.93(d,J=1.9 Hz, 1H), 7.66~7.62(dd,J=7.3 Hz, 1.8 Hz, 1H),7.37~7.22(m, 5H), 6.98~6.94(d,J=7.3 Hz, 1H), 5.59(s, 1H), 3.34~3.25(m, 2H), 2.69~2.65(m, 2H), 1.95(s, 1H); MS(ESI)m/z: 283.1{[M+H]+}。
1d: m.p.134~136 ℃;1H NMRδ: 11.68(s, 1H), 7.08~7.04(d,J=6.7 Hz, 1H), 7.92~7.88(dd,J=6.7 Hz, 1.9 Hz, 1H), 6.66~6.65(d,J=1.9 Hz, 1H), 3.84~3.82(m, 1H), 3.83(s, 3H), 3.31~3.23(m, 2H), 2.65~2.63(m, 2H), 1.95(s, 1H), 1.72~1.69(m, 2H), 0.97~0.95(t,J=13.6 Hz, 3H); MS(ESI)m/z: 231.1{[M+H]+}。
(2)2a~2d的合成(以2a為例)
將1a248 mg(1.0 mmol)溶于20.0 mL四氫呋喃,加入DDQ 1 g(4.4 mmol),室溫攪拌24 h。減壓蒸除溶劑,剩余物用50.0 mL乙酸乙酯溶解后經飽和碳酸鈉水溶液和飽和食鹽水洗滌,無水硫酸鈉干燥。過濾后將濾液減壓濃縮,殘余物經硅膠柱層析(洗脫劑:V二氯甲烷∶V甲醇∶V氨水=30.0 ∶1.0 ∶0.2)純化得灰白色粉末2a158 mg,收率65%。以類似方法合成灰白色粉末2b~2d。
2a: m.p.244~246 ℃(m.p.245~246 ℃[19]);1H NMRδ: 11.72(s, 1H), 8.39~8.37(d,J=7.1 Hz, 1H), 8.21~8.19(d,J=6.6 Hz, 1H), 7.69~7.65(m, 2H), 7.54~7.51(m, 2H), 7.32~7.22(m, 5H); MS(ESI)m/z: 245.1{[M+H]+}。
2b: m.p.260~262 ℃(分解);1H NMRδ: 11.71(s,1H), 8.38~8.36(d,J=7.1 Hz, 1H), 7.72~7.69(m, 2H), 7.53~7.49(m, 2H), 7.28~7.21(m, 4H), 7.05~7.03(d,J=6.7 Hz, 1H), 3.83(s, 3H); MS(ESI)m/z: 275.1{[M+H]+}。
2c: m.p.273~275 ℃(分解);1H NMRδ: 11.73(s, 1H), 8.39~8.37(d,J=7.1 Hz, 1H), 7.71~7.68(d,J=7.1 Hz, 1H), 7.57~7.51(m, 3H), 7.31~7.19(m, 5H); MS(ESI)m/z: 279.1{[M+H]+}。
2d: m.p.231~233 ℃;1H NMRδ: 11.72(s, 1H), 8.67~8.65(d,J=7.1 Hz, 1H), 7.93~7.91(d,J=7.1 Hz, 1H), 7.73~7.72(d,J=1.8 Hz, 1H), 7.53~7.51(d,J=8.3 Hz, 1H), 7.06~7.03(d,J=8.3 Hz, 1H), 3.83(s, 3H), 3.46~3.42(q,J=13.1 Hz, 2H), 1.34~1.32(t,J=13.1 Hz, 3H); MS(ESI)m/z: 227.1{[M+H]+}。
2 結果與討論
2.1 1a的合成
以1a的合成為例,研究催化劑用量、溶劑、反應溫度和反應時間對1a收率的影響,結果見表1。由表1可以看出,催化劑六氟異丙醇鈣的用量為0.2 eq.時最佳,增大用量至0.5 eq.不能明顯增加收率(Entry 1)。當用量為0.1 eq.時,延長反應時間,色胺也難以轉化完全,且收率降低(Entry 3)。苯甲醛用量在1.5 eq.時效果最佳,收率為83%(Entry 4)。增加苯甲醛用量對收率影響不大,降低至1.2 eq.時會導致收率降低(Entry 5)。由表1還可以看出,同等條件下使用DCM作溶劑相較于四氫呋喃(THF)效果較好(Entry 4, Entry 6),這可能與反應物溶解性或溶劑極性有關;使用THF或二氯乙烷作溶劑,升高反應溫度(Entry 7~8),均使收率降低,且反應中發現有較多副產物生成。因此,最佳的反應條件(Entry 4)為:色胺2.0 mmol,苯甲醛3.0 mmol,六氟異丙醇鈣0.4 mmol, DCM 20.0 mL,于室溫下反應24 h。
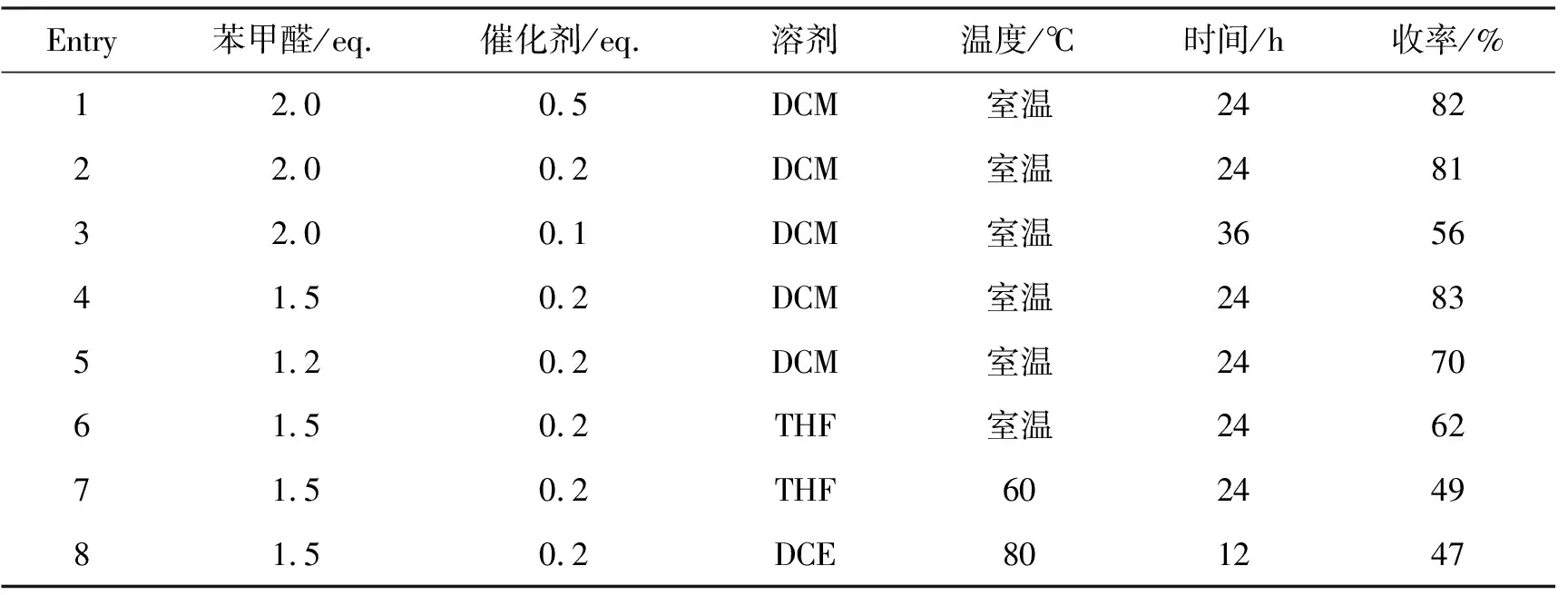
表1 1a的合成條件優化*
2.2 Pictet-Spengler中的取代基效應
為考察取代基效應,利用1a合成中確定的最優反應條件,在色胺底物中引入甲氧基、氯取代基,并將苯甲醛擴展至脂肪族醛類,合成多取代四氫-β-咔啉化合物(1b~1d),對比結果如表2所示。由表2分析可以發現(1a~1c),當底物色胺的芳香環上存在甲氧基等斥電子取代基時,收率較高;當存在氯等吸電子基時,收率會相對降低,說明Pictet-Spengler反應中底物色胺存在取代基效應,這可能是由于芳香環上的電子云密度增加有利于π電子進攻亞胺中間體,從而促進反應進行。同時比較1b與1d,發現使用芳香醛時收率顯著大于脂肪醛,可能是由于芳香醛能夠與色胺生成更加穩定的亞胺中間體,降低了反應活化能。
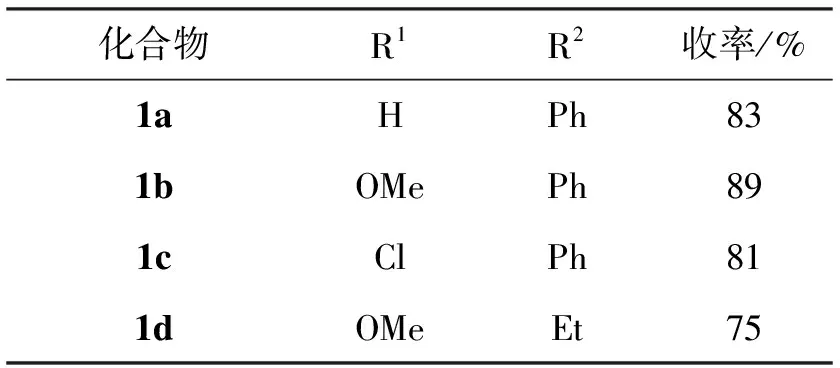
表2 不同取代基對Pictet-Spengler反應中收率的影響
2.3 2a~2d合成中底物結構影響
在2a~2d的合成中,采用經典的DDQ氧化法對四氫-β-咔啉化合物(1a~1d)脫氫芳香化,發現反應中同樣存在取代基效應,對比結果如表3所示。從表3可以看出,在氧化脫氫反應中,底物1a~1c吲哚環上無論是吸電子基還是斥電子基都能使收率略微提高(2a~2c)。而當底物1d哌啶環上取代基為脂肪烴基時,收率顯著小于芳香烴基的底物,可能是由于芳香烴基的共軛作用促進了反應進行。
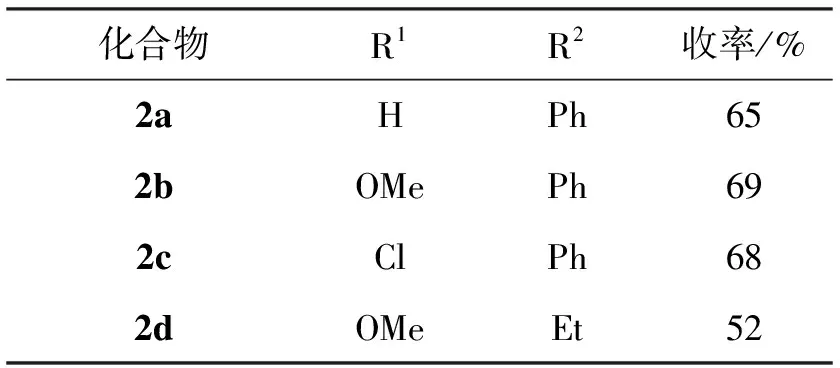
表3 不同取代基對氧化脫氫反應中收率的影響
3 結論
本文以取代色胺和醛類為底物,0.2 eq.六氟異丙醇鈣為催化劑,DCM為溶劑,室溫反應24 h;再經過經典DDQ氧化脫氫反應合成了4個多取代β-咔啉類目標化合物,并考察了反應適用性與取代基效應。該方法避免了傳統Pictet-Spengler反應路線中質子酸的使用,適用于在非酸性條件下合成含有對酸敏感基團的多取代β-咔啉類化合物。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
